Epileptic Disorders
MENULocation-atypical lesions in non-ketotic hyperglycemic epilepsy: expanding the clinico-radiographic phenotype Volume 24, issue 6, December 2022

Non-ketotic hyperglycemia (NKH) can present with a spectrum of neurological disease attributed to cytotoxic neuronal injury in patients with poorly controlled diabetes. In cases in which cortical and juxtacortical areas are involved, seizures commonly manifest. Several magnetic resonance imaging (MRI) studies on NKH-associated seizures (NKHS) have described subcortical T2-based hypointensity, leptomeningeal enhancement, and gyral T2-based hyperintensity [1], typically affecting the occipital and posterior parietal cortex, and characteristically associated with hemianopsia [2, 3]. Here, we report four patients with acute presentation of NKHS who had typical MRI findings in atypical brain locations.
Patient 1
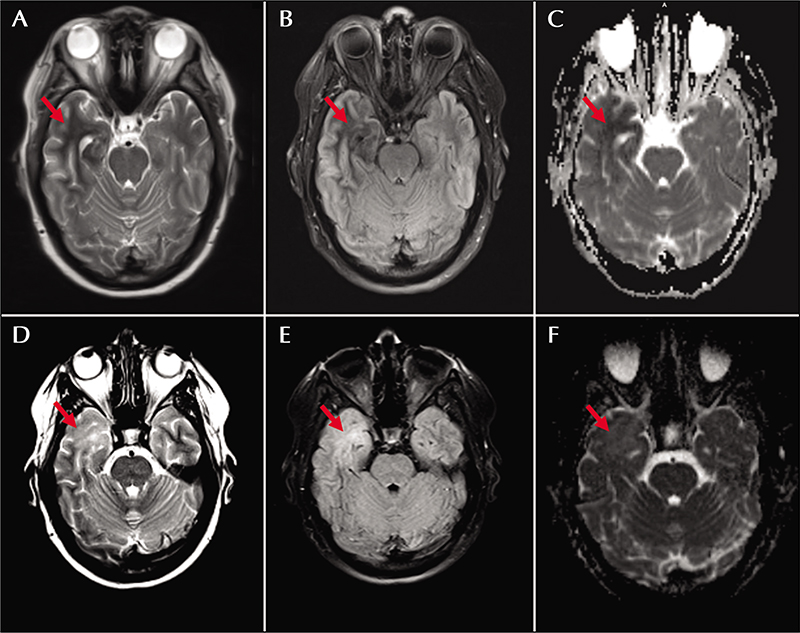
A 47-year-old woman with type 2 diabetes mellitus and idiopathic intracranial hypertension (IIH) presented with 90-second episodes of burning dysesthesias over her left face and arm evolving to lip smacking, left arm elevation, and decreased responsiveness. On presentation, brain MRI showed a T2/FLAIR hypointense area within the right anterior temporal white matter, which exhibited restricted diffusion and involved both MCA and ACA territories of the anterior temporal lobe (figure 1A-C). Laboratory testing showed serum glucose at 627 mg/dL without anion gap (14 mmol/L) and hemoglobin A1c (Hgb A1c) at 16.1%. Serum sodium was 127 mmol/L (140 mEq/L when corrected for glucose, per the Hillier correction [4]); serum electrolytes were otherwise within normal limits and urine ketones were negative. Cerebrospinal fluid (CSF) analysis showed mild hyperglycorrhachia (119 mg/dL), but otherwise no evidence of an immune, inflammatory or infectious process. Continuous video electroencephalography (CV-EEG) captured two electrographic seizures that localized to the right temporal lobe. Correction of hyperglycemia, initiation of levetiracetam (1500 mg twice daily), and continuation of topiramate (100 mg twice daily, prescribed for IIH), resulted in interval resolution of her symptoms.
The patient remained adherent to an insulin-based diabetic regimen following discharge. Her levetiracetam was also lowered to 1000 mg twice daily and topiramate continued at her prior dosage. Follow-up imaging at 7.5 weeks showed interval resolution of the previously noted T2/FLAIR hypointense areas within the right anterior temporal lobe with interval development of T2/FLAIR hyperintensity within this region. There was normalization of the ADC map without associated atrophy. The findings were deemed compatible with gliotic evolution of the patient’s initial insult (figure 1D-F). She was continued on antiepileptics for another year before ultimately discontinuing without further events.
Patient 2
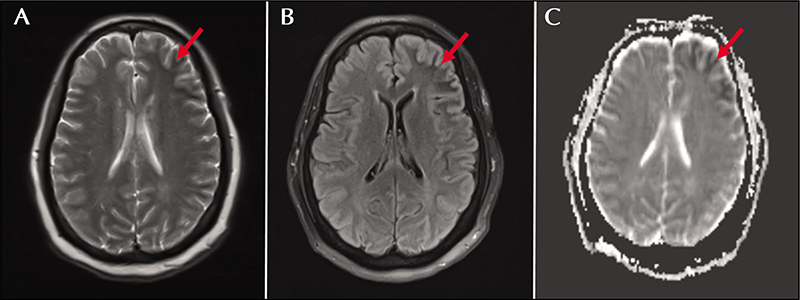
A 56-year-old woman with type 2 diabetes mellitus, hypertension, and peripheral vascular disease presented with a two-week history of recurrent episodes of behavioral arrest. She had a witnessed generalized tonic-clonic seizure on arrival to the emergency department. Brain MRI showed juxtacortical white matter T2/FLAIR hypointensity in the left frontal and temporal lobes (figure 2). Laboratory testing showed a glucose level of 447 mg/dL with associated anion gap metabolic acidosis (16 mmol/L) and elevated Hgb A1c (12.4%). Urine ketones were negative and her metabolic acidosis was attributed to recent seizure activity, given rapid correction following cessation of seizure activity. She was started on levetiracetam, 1000 mg twice daily. While hospitalized, she experienced numerous 1-2-minute episodes of sudden speech arrest with inattention and inability to follow commands. A routine EEG captured left frontal seizures. Fourteen additional left frontal seizures were captured on CV-EEG. Levetiracetam was increased to 2,000 mg twice daily and oxcarbazepine at 300 mg twice daily was added. This regimen, in conjunction with glucose control, prevented further seizures. The patient was lost to follow-up, precluding repeat brain MRI.
Patient 3
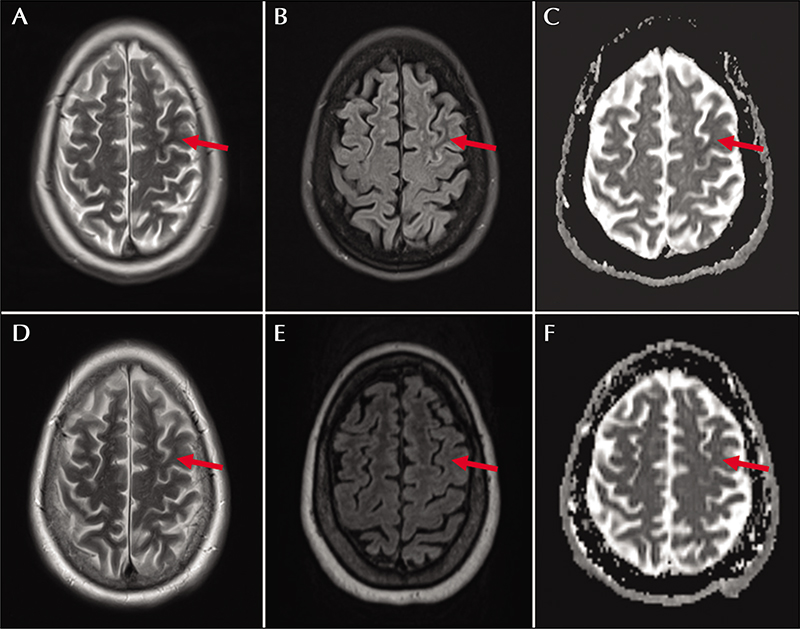
A 60-year-old woman with type 2 diabetes mellitus, hypertension, and prior right cerebellar ischemic stroke presented with two days of intermittent spells of “confusion” and transient forgetfulness. Her interictal exam was notable for subtle paraphasic errors in speech and mild right hemiparesis, confirmed as new deficits by collateral sources. Brain MRI showed a subtle T2/FLAIR hypointense area in the left precentral gyrus with corresponding diffusion restriction (figure 3). Laboratory testing showed glucose at 548 mg/dL without anion gap (14 mmol/L) or ketosis. Her Hgb A1c was 15.5% (7.8% one year prior). Serum sodium was 127 mmol/L (140 mEq/L corrected). CSF analysis showed hyperglycorrhachia (115 mg/dL), but was otherwise unremarkable. Two electrographic seizures originating from the left centro-parietal region were captured by CV-EEG over several days of monitoring. Semiology of the first was rhythmic right foot movement while the second was characterized by right hand tapping. Her seizures were controlled with levetiracetam, 750 mg twice daily, and normalization of her glucose. Serum sodium normalized with euglycemia. She returned to her prior baseline over several days. She remained closely adherent to an insulin-based diabetic regimen and remained seizure-free at follow-up, nine months later. Repeat imaging at that time showed nearcomplete resolution of the left middle central gyrus hyperintensity and corresponding diffusion restriction (figure 3D-F). Antiepileptics were discontinued at follow-up without further events.
Patient 4
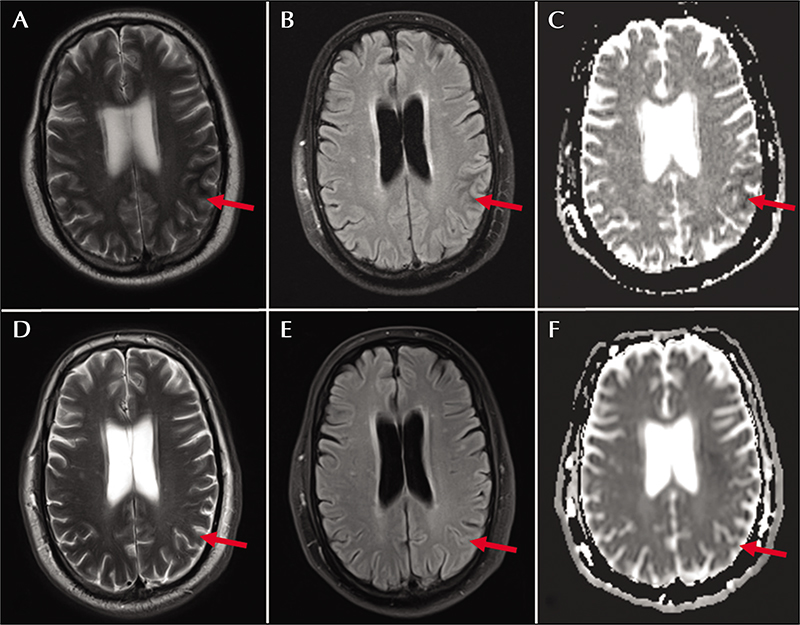
A 57-year-old man with type 2 diabetes mellitus and coronary artery disease presented with a three-week history of episodic right-hand clenching and twitching. The episodes were initially 30 seconds in duration but had progressed over a three-week period, and at the time of presentation were lasting up to 10 minutes. Brain MRI showed an area of subtle cortical thickening with T2/FLAIR hypointensity in the left parietal lobe (figure 4A-C). Laboratory testing was notable for glucose at 527 mg/dL with mild anion gap (18 mmol/L) and Hgb A1c at 10.9%. Serum electrolytes were within normal reference ranges and urine ketones were not detected. The hand movements exhibited an electrographic correlate, and numerous electrographic seizures originating from the left centro-parietal region were captured on CV-EEG. His seizures were wellcontrolled with levetiracetam, 1,500 mg twice daily, and correction of his hyperglycemia. He remained adherent to an insulin-based diabetic regimen and remained seizure-free at follow-up, seven months later. Repeat imaging at that time showed near-complete resolution of the left parietal lobe T2/FLAIR hypointensity, no contrast enhancement, and mild residual atrophy (figure 4D-F). His antiepileptics were discontinued without further events.
Discussion
The neurologic manifestations of sustained hyperglycemia are protean. Classically associated with hemichorea-hemiballism (HC-HB) when affecting the basal ganglia [5], NKH has also been associated with myoclonus, opsoclonus, and seizures [6, 7]. Seizures in the setting of hyperglycemia were first reported by Maccario et al. in 1965 [8]. While patients can show some response to antiepileptic medication, seizure freedom is typically not achieved until the hyperglycemia is corrected [9]. Here, we present a series of four patients with NKH in the setting of poorly controlled diabetes with neurologic signs corresponding to focal onset seizures, each with appropriately localizing T2/FLAIR subcortical hypointensity in the frontal, upper parietal and temporal lobes. Similar findings have been well described in the occipital and posterior parietal lobes in the context of NKH, which can characteristically lead to hemianopsia that may recover upon treatment of the NKH.
Several studies have characterized the MR findings of NKHS, which most commonly include subcortical T2-based changes [10] and gyral swelling [10, 11]. T2 features are most frequent and include T2 hypointensity or isointensity in areas of abnormal T1 signal [11-13]. Some studies have shown variable levels of restricted diffusion corresponding to the areas of these structural MRI abnormalities, as demonstrated by our cases [11, 12]. Furthermore, while the T2-weighted hypointensity and diffusion restriction often normalize over a period of weeks with correction of the hyperglycemia (as seen in Patients 3 and 4), they may also reflect gliosis, evolving into a hyperintense T2-weighted signal (as seen in Patient 1) [14].
It remains unclear why NKH results in localized regions of T2 hypointensity. Subcortical T2 hypointensity is not unique to NKH, and has been reported in the context of ischemia, revascularization, encephalitis and meningitis, leptomeningeal metastasis, focal seizures and Sturge-Weber syndrome [15-18]. Several hypotheses have been put forward for this often transient subcortical T2 hypointensity, including free radical formation, iron deposition, and venous congestion [16, 19]. Another potential explanation for these imaging findings could be a focal loss of free water, which would be expected to both decrease the T2 signal and diffusion. Indeed, the converse -an increase in free water, i.e., edema- is associated with both an increase in T2 signal and ADC. Application of advanced diffusion imaging, such as diffusion basis spectrum imaging, might help to investigate this possibility further.
The imaging findings in NKHS are usually noted within posterior and precentral cortical areas [20, 21]. Lee et al. reported the largest neuroimaging series of NKHS to date and found that nine patients had involvement of the parieto-occipital region with two patients having precentral gyrus abnormalities [21]. Accordingly, these patients manifested with visual obscurations and hallucinations, reflecting parietooccipital involvement. The “hyperglycemic hemianopia syndrome” has also been described, implicating frequent visual seizures originating from the occipital lobes, with corresponding subcortical T2 hypointensity in these areas [22, 23]. Our cases are intended to raise awareness amongst neurologists and radiologists, that “anterior” cortical regions of the brain might be similarly involved in the context of NKH.
The phenomena observed in our case series, with burning dysesthesias, lip smacking (Patient 1), generalized tonic-clonic activity (Patient 2), right arm and leg shaking (Patient 3), and right-hand clenching (Patient 4) offer localizing value to the temporal lobes, bilateral hemispheres, frontal cortex, and hand knob, respectively (table 1) [24]. Findings on brain MRI and CV-EEG corroborate these localizations. In the three cases with available follow-up imaging, these imaging abnormalities corrected with resolution of hyperglycemia (figures 1, 3-4). Cases of NKHS involving these atypical brain regions provide an important addition to the myriad manifestations of hyperglycemia.
Alternative diagnoses were considered. Hyponatremia itself, which was observed in two of our patients, can cause seizures [25], but is not associated with focal areas of T2 hypointensity. Rather, the mild hyponatremia was likely due to the relative dilutional effect of the hyperosmolar state, as seen when values were adjusted per the Hillier correction factor [4]. Infarcts can also be associated with subcortical T2 hypointensity, but the clinical course in these four patients argues against this differential diagnosis. Moreover, the imaging features were not typical of infarct, including, in one case, involvement of two adjacent vascular territories. Autoimmune encephalitides, such as those associated with antibodies directed against leucine-rich glioma-inactivated 1 (anti-Lgi1), can present with a prodrome of hyponatremia (via the syndrome of inappropriate diuretic hormone) and T2-based MR changes, though these are typically hyperintense with strong predilection for the medial temporal lobes [26].
Our case series demonstrates that NKH-related seizures can be associated with typical MRI features in an atypical distribution, specifically in anterior regions of the brain that can be localized by seizure semiology.
Supplementary material
Summary slides accompanying the manuscript are available at www.epilepticdisorders.com.