Epileptic Disorders
MENUAcute symptomatic seizures: an educational, evidence-based review Volume 24, issue 1, February 2022



Epileptic seizures occur in all persons with epilepsy but not everyone who experiences an epileptic seizure suffers from epilepsy. Indeed, up to 40% of all epileptic seizures occur in people with an acute brain insult but without epilepsy [1]. These seizures are defined as acute symptomatic seizures. There are many causes for acute symptomatic seizures, which all alter the excitability of the central nervous system (CNS), leading to a transient lowering of the seizure threshold (figure 1). In contrast to epilepsy, based on the general concept of acute symptomatic seizures, seizures are not expected to recur once the precipitating factor or condition has been removed or reversed. However, in practice, as we will later see, an increased risk for the development of epilepsy exists after acute symptomatic seizures caused by structural brain pathologies. Awareness of the potential of certain CNS pathologies to cause acute symptomatic seizures is important to provide prompt treatment. Moreover, the accurate distinction between an acute symptomatic seizure and an unprovoked seizure provides a different prognosis and alters treatment. This review aims to provide an educational overview about the definition, epidemiology, causes, management and prognosis of acute symptomatic seizures. Special emphasis is given to the learning objective listed in the educational curriculum for epileptologists which was recently published by the International League against Epilepsy [2]: Accurately distinguish acute symptomatic seizures from epilepsy.
Definitions of an epileptic seizure and epilepsy
Before we can discuss about what is an acute symptomatic seizure, a quick review of the definitions of an epileptic seizure and epilepsy in general is necessary.
An epileptic seizure has been defined as the “transient occurrence of signs and/or symptoms due to an abnormal excessive or synchronous activity in the brain” [3].
Epilepsy is a brain disorder with various and heterogeneous causes of which the main manifestation is epileptic seizures which in turn effect people's behaviour, personality and life. The hallmark of epilepsy is the recurrence of epileptic seizures.
The International League against Epilepsy (ILAE) has proposed a conceptual definition of epilepsy as a brain disorder “that is characterised by an enduring predisposition of the brain to generate epileptic seizures and by the neurobiological, cognitive, psychological and social consequences of this condition” [3].
Any definition of epilepsy usually requires that the epileptic seizures which are taken into account occur in the absence of any apparent precipitating factors or conditions, i.e. that they are “unprovoked” [4].
The cause of unprovoked seizures may be a CNS insult in the remote past. The length of the time between the brain insult and seizures in the absence of ongoing active disruption of CNS integrity determines that the seizure is unprovoked or “remote symptomatic”.
On that basis, the clinical or operational definition of epilepsy from the ILAE states that epilepsy exists in a person who had “at least two unprovoked seizures more than 24 hours apart or who had one unprovoked seizure and has a probability for the recurrence of further seizures that is similar to the recurrence risk after two unprovoked seizures (that is at least 60%) over the next 10 years” [4].
The conceptual definition of epilepsy supported by the ILAE differs from this clinical or operational definition [3]. The conceptual definition of epilepsy does not necessarily require a seizure to be unprovoked but requires an enduring predisposition of the brain to potentially generate further seizures. As an example, reflex epilepsies fulfil the conceptual definition of epilepsy even though all seizures are provoked, because there is an enduring alteration in the brain to generate seizures in response to the stimulus.
Definition of an acute symptomatic seizure
An acute symptomatic seizure “occurs in close temporal relationship with an acute CNS insult, which may be metabolic, toxic, structural, infectious, or due to inflammation” [5].
Acute symptomatic seizures are different from unprovoked seizures in several aspects and are therefore not included in the definition of epilepsy.
First, unlike in unprovoked seizures, there should always be a clearly identifiable, concomitant acute, causal condition that has occurred close to the time of the seizure. The cause may be an acute disturbance of structural brain integrity such as a cortical haemorrhage, or a disturbance of brain function due to, e.g. alcohol withdrawal, present at the time of the seizure.
Secondly, acute symptomatic seizures usually do not recur once the precipitating factor or condition has been removed or reversed and the functional integrity of the CNS has been restored. This is in contrast to epilepsy where seizures are expected to recur. The lack of an “enduring predisposition” after an acute symptomatic seizure means epilepsy does not exist. For example, if a patient has two epileptic seizures due to severe hyponatraemia, there is no “enduring predisposition” once the hyponatraemia has resolved. However, the distinction between acute symptomatic seizures and epilepsy is more complicated in persons with acute symptomatic seizures due to destructive brain pathologies, such as stroke or head trauma, because they have an increased risk for the later development of epilepsy (discussed below).
What is thought to be a “close temporal relationship” between a CNS insult and a seizure varies according to the underlying pathology. For example, a seizure is considered acute symptomatic if it occurs within the first seven days of a stroke or a traumatic brain injury [5]. In other conditions, an acute symptomatic seizure may occur longer than a week after the onset of the brain insult provided that there is evidence of a continued active brain disease. An example is acute inflammatory CNS disease (e.g. infectious or autoimmune encephalitis). For other conditions, a closer temporal relationship is required to prove a plausible causality, e.g. disorders such as hyponatraemia, where there must be evidence of low serum sodium level within 24 hours of the seizure.
Seizures that are the manifestation of a neurodegenerative disease such as Alzheimer's dementia may be referred to as progressive symptomatic seizures. They are not acute symptomatic seizures as the cause of the seizure is neither transient nor reversible but a persistent and progressive condition. In such cases, a diagnosis of epilepsy can definitively be made after a second seizure but might be made even after the first seizure, if there is evidence of a recurrence risk greater than 60%. Similarly, seizures that arise from most brain tumours are progressive symptomatic seizures, unless the brain tumour can be fully resected and seizures disappear. For example, in a patient who presents with a first seizure as the first manifestation of a meningioma, the seizure could be rated as acute symptomatic if the tumour is completely removed and there are no further seizures. Here, the definition of acute symptomatic can only be made in retrospect. In multiple sclerosis, a seizure should be considered acute symptomatic if it occurs on presentation or within seven days of a relapse [5].
Febrile seizures are defined as any seizure that occurs in association with fever >38̊C in a child aged about six months to five years of age without evidence of CNS infection. They are a classic form of acute symptomatic seizure and are the most common type of epileptic seizures with a lifetime prevalence of 2-6% in the overall population. Because they represent a rather distinct entity and are specific to the paediatric population, they will not be discussed further in this text. For a comprehensive review on febrile seizures, please see [6].
In clinical practice, several terms, which are similar to the term “acute symptomatic seizure”, such as “provoked seizure”, “situation-related seizure” and “reactive seizure”, are frequently used. The ILAE has proposed that these terms are synonymous with and should be replaced by acute symptomatic seizure [5].
Critical issues and potential pitfalls in defining acute symptomatic seizures
The main problem in the definition of acute symptomatic seizures arises from the difficulty in combining, in a single concept, both seizures that are caused by acute structural brain pathologies and seizures that are caused by provocative factors. It has been argued that seizures caused by an acute structural brain lesion, such as a stroke, should not be equalled under one term to seizures provoked by a truly reversible factor such as hyponatraemia [7]. The 10-year risk for the recurrence of an unprovoked seizure after an acute symptomatic seizure due to a stroke was found to be 33% [8]. This is substantial, but still means that an acute symptomatic seizure alone does not qualify as epilepsy. However, if one specifies for stroke subtype, then the risk is even higher, e.g. as predicted by the SeLECT score [9]. On the other hand, acute symptomatic seizures caused by a reversible factor or condition, such as an intoxication or hyponatraemia, are assumed to be linked to a very low risk for further unprovoked seizures, although precise data for recurrence risk is lacking.
The distinction between “provoked” or “unprovoked” can be challenging. It is difficult to absolutely exclude a provocative factor even if a seizure appears to be unprovoked. On the other hand, the presence of a potentially provocative factor does not exclude the existence of an underlying predisposition for the generation of epileptic seizures [4].
In some situations, such as the occurrence of a seizure in the immediate context of alcohol withdrawal or hyponatraemia, the seizure event will be confidently judged as provoked and will not result in a diagnosis of epilepsy. The situation is less clear in the context of sleep deprivation. Extensive sleep deprivation could potentially provoke a seizure in an individual without any underlying predisposition for the development of seizures, however, sleep deprivation is also well established as a typical provocative factor in idiopathic generalised epilepsies.
Furthermore, the exclusive presence of provoked seizures does not mean that epilepsy does not exist; as noted above, provoking factors may be present with every seizure in people with reflex epilepsies – here there is an enduring abnormal alteration of brain function meeting at least the conceptual definition of epilepsy [3].
The definition of a ”close temporal relationship” as well as the proposed cut-off levels of laboratory values for acute symptomatic seizures due to metabolic derangements have attracted criticism due to the fact that they are relatively arbitrary and not backed by clear data [7]. For example, in electrolyte disturbances, the acuteness in change seems to be more important for seizure risk than the change in absolute levels [10]. If a seizure is suspected to be caused by a metabolic derangement but ILAE cut-off levels of laboratory values are not met, it has been recommended not to label the seizure as acute symptomatic [5]. However, this does not mean that the seizure can be conversely called an unprovoked seizure. In such a case, the relationship with a metabolic derangement should be treated as “unknown” and the seizure should not be rated as epilepsy.
Incidence of acute symptomatic seizures
It has been estimated that acute symptomatic seizures account for up to 40% of all epileptic seizures [1]. In Rochester, Minnesota, over a 50-year period, acute symptomatic seizures accounted for 34% of all epileptic seizures [11].
These estimates are largely based on data from higher-income countries and it is possible that in other geographical regions, where CNS infections are endemic and head trauma more common, acute symptomatic seizures will be even more common.
It has been pointed out that not many epidemiological studies have been conducted on acute symptomatic seizures because of various difficulties:
- •often, only the underlying aetiology but not the seizure will be indexed as a diagnosis, complicating any retrospective review;
- •some patients with acute symptomatic seizures will not be further evaluated or lost to follow-up (e.g. alcohol withdrawal seizures);
- •in addition, studies on seizure epidemiology have often failed to distinguish between acute symptomatic and unprovoked seizures in the past [1].
In a retrospective study that specifically investigated the frequency of acute symptomatic seizures in a large population in the United States over a 50-year period, the annual incidence was 39/100,000 [12]. Smaller studies have reported similar incidence rates for acute symptomatic seizures [13-15].
The incidence of acute symptomatic seizures is significantly lower compared to the incidence of unprovoked seizures for which incidence rates vary between 42-61/100,000/year [1].
Overall, acute symptomatic seizures are more common in male than in female individuals. An age-adjusted incidence rate of 42/100,000/year was reported in male individuals compared to an incidence rate of 27/100,000/year in females [12]. The lifetime risk for the occurrence of an acute symptomatic seizure was calculated as 5% for males compared to 2.7% for females [12].
Comparable to epilepsy, the age-specific incidence rate for acute symptomatic seizures is highest in the first year of life, decreases in childhood and early adulthood and thereafter starts increasing with age, culminating in a second peak in individuals aged 80 years and older [12].
Age and sex patterns regarding the incidence of acute symptomatic seizures correlate with the age and sex distributions of the causative pathologies. The high incidence of acute symptomatic seizures in the first year of life is associated with the relatively frequent occurrence of encephalopathies (e.g. hypoxic-ischaemic encephalopathy), CNS infections, vascular aetiologies and metabolic disorders in the neonatal period. Traumatic brain injuries, which occur predominantly in younger men, are the most common cause of acute symptomatic seizures in early adulthood. Alcohol and drug withdrawal is another common cause of acute symptomatic seizures that is more frequently seen in younger and middle-aged men. Cerebrovascular disease is the most common cause of acute symptomatic seizures in elderly patients. The rising incidence of cerebrovascular disease with age is largely responsible for the increase in incidence of acute symptomatic seizures in older individuals [12].
Mortality and acute symptomatic seizures
Patients who suffer from acute symptomatic seizures have a high risk for mortality in the weeks after the event. A study of two independent cohorts of patients with acute symptomatic seizures determined a case fatality rate of 20% in the first 30 days after the seizure [16]. The risk of death was significantly higher in older individuals (aged 65 years or older) than in younger individuals. Cerebrovascular disease and hypoxic encephalopathy were determined as the predominate causes of acute symptomatic seizures in those patients who had a fatal outcome in the first 30 days after the seizure.
In a study of hospitalised patients, those who had a seizure for the first time in their life, most of them due to an acute symptomatic cause, had a significantly higher chance of a negative outcome (death or discharge to a hospice) than patients who already had a history of seizures prior to hospitalisation [17]. Cerebrovascular disease was found to be the most frequent aetiology of seizures in these patients, followed by metabolic disturbances and brain tumours.
Another study compared the mortality of patients who had an acute symptomatic seizure with patients who had a first unprovoked seizure. Those with acute symptomatic seizures had an 8.9-fold higher mortality rate in the first 30 days after the seizure event [8]. No difference in mortality was evident after 10 years of follow-up between these two groups.
Mortality is particularly high in patients with acute symptomatic status epilepticus (SE). In a study of 184 patients with both acute symptomatic and unprovoked SE, 89% of deaths (n=38) in the first 30 days after the event occurred in patients with acute symptomatic SE [18]. This accounted for a case fatality rate of 34% for patients with acute symptomatic SE compared with 5% with unprovoked SE. Age older than 65 years and male gender were significantly associated with a higher risk of death, and the majority of those patients who died after acute symptomatic SE suffered either from cerebrovascular disease or hypoxic encephalopathy. Acute symptomatic SE is also associated with an increased risk of long-term mortality [19].
The high mortality in patients with acute symptomatic seizures is largely attributed to the underlying causative brain pathologies [16]. Common aetiologies of acute symptomatic seizures such as cerebrovascular disease, brain tumours or hypoxic encephalopathy are well known to entail a high risk of mortality, whether or not they occur in conjunction with a seizure. The influence of acute symptomatic seizures on the clinical outcome of patients with acute brain diseases is unclear. The occurrence of an acute symptomatic seizure may simply reflect the severity of the underlying acute brain insult.
Whether acute symptomatic seizures themselves contribute negatively to a patient's clinical outcome has been debated. This issue has been particularly studied in patients with stroke and some studies have demonstrated that acute symptomatic seizures are independently associated with higher mortality [20] while others have failed to show such an effect [21]. However, a recent, large, retrospective analysis of 1,787 patients with acute symptomatic seizures in the setting of acute ischaemic stroke controlled for stroke severity using the National Institutes of Health Stroke Scale (NIHSS). Those with acute symptomatic seizures had almost twice the risk of in-hospital death compared to those without seizures, suggesting that at least for ischaemic stroke, acute symptomatic seizures might indeed negatively influence a patient's outcome independent of disease severity [22].
Risk of seizure recurrence after acute symptomatic and unprovoked seizures
The recurrence of unprovoked epileptic seizures is a defining characteristic of epilepsy. Yet five years after a single unprovoked seizure, the risk of seizure recurrence lies only between 40%-50% [23]. However, the recurrence risk is much higher after a second unprovoked seizure. In one study of adults, an individual with a second unprovoked seizure had a 73% risk for a third unprovoked seizure and an individual with a third unprovoked seizure had a 78% risk for a fourth [24]. The risk of recurrence after a first unprovoked seizure is increased by a remote symptomatic cause, epileptiform EEG abnormalities, abnormal brain imaging and nocturnal seizures [25].
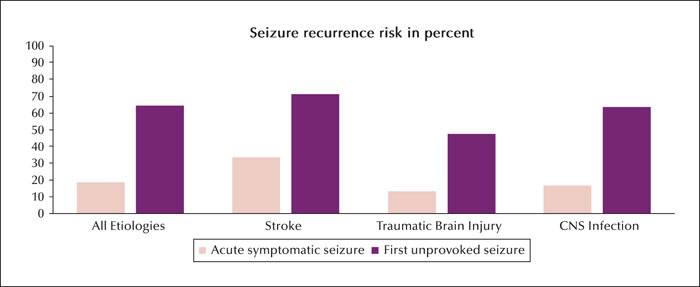
The theoretical concept and definition of an acute symptomatic seizure suggests that the seizure recurrence risk should be relatively low. Evidence to support this hypothesis is provided by the landmark study of Hesdorffer et al., which retrospectively compared the risk of seizure recurrence between 262 patients with acute symptomatic seizures and 148 patients with a first unprovoked seizure [8]. For better comparison, seizure aetiologies in both groups, either acute or remote symptomatic, respectively, were confined to the following three brain pathologies: stroke, traumatic brain injury and CNS infection. For each of the three aetiologies, patients with acute symptomatic seizures had a significantly lower risk (80% relative risk reduction) of developing a subsequent seizure compared to patients with a first unprovoked seizure (overall 18.7% vs. 64.8%) during a 10-year follow-up (figure 2). The clear differences in seizure recurrence between acute symptomatic seizures and first unprovoked seizures revealed by this study strongly support the theoretical assumption that most acute symptomatic seizures are not epilepsy as they are “not associated with an enduring predisposition to generate seizures” [8]. However, the risk of subsequent seizures after an acute symptomatic seizure is substantial (33% for stroke, 13.4% for traumatic brain injury and 16.6% for CNS infection) (see figure 2) and almost approaches the recurrence risk for another unprovoked seizure after a first unprovoked seizure without underlying structural CNS disease [25]. In certain situations, e.g. if one specifies for stroke subtype and includes more risk factors, then the risk of further seizures after a stroke that occurred with an acute symptomatic seizure is even greater, e.g. as predicted in the SeLect score [9]. These substantial seizure recurrence risks are probably related to the destructive nature of the three brain pathologies and similar recurrence risks would not be expected following acute symptomatic seizures from causes such as hyponatraemia. However, data on seizure recurrence risk after acute symptomatic seizures due to reversible factors is lacking. It has been pointed out that it would be difficult to carry out similar studies to compare the recurrence risk of acute symptomatic seizures caused by metabolic derangements relative to first unprovoked seizures [8].
Specific causes of acute symptomatic seizures
In adults, the most common aetiologies of acute symptomatic seizures are cerebrovascular disease, traumatic brain injury, drug and alcohol withdrawal, and CNS infections, with each aetiology accounting for about the same proportion of cases (figure 1). Less common causes of acute symptomatic seizures include metabolic disorders, encephalopathy, intoxications and eclampsia [12]. Table 1 presents the risks and risk factors for acute symptomatic seizures regarding the most important aetiologies (table 1).
Cerebrovascular causes
The estimated frequency of acute symptomatic seizures after stroke is 3-6%. The rate of seizures is significantly higher in patients with haemorrhagic (10-18%) rather than ischaemic stroke (2-4%) [26]. Lobar intracerebral haemorrhage, subarachnoid haemorrhage, and ischaemic stroke with secondary haemorrhagic transformation show a stronger association with acute symptomatic seizures than ischaemic stroke [26]. Although ischaemic stroke is associated with a lower risk in comparison to intracerebral haemorrhage, it accounts for a larger overall burden of post-stroke seizures because of its higher incidence, and represents the leading aetiology of acute symptomatic seizures in the elderly.
Acute symptomatic seizures generally occur during the first one or two days after cerebral ischaemia, with about two thirds within the first 24 hours. Most seizures associated with haemorrhagic stroke occur at the onset or within the first 24 hours. The majority of seizures are focal, regardless of stroke type or time of seizure occurrence. This is not surprising given the accepted hypothesis that the focal damage following stroke can act as the focus of epileptic activity and a fast, bilateral spread from a focal seizure generator cannot be ruled out in the case of generalized seizures [27].
Acute symptomatic seizures after stroke are thought to be the result of acute biochemical dysfunction and excitatory neurotransmitter release leading to transient changes in neuronal excitability and electrically irritable tissue [28]. Pro-excitatory cellular changes follow acute ischaemic neuronal injury and include accumulation of intracellular calcium and sodium and increased extracellular concentrations of glutamate, which may lead to depolarization of the transmembrane potential and lower the seizure threshold [28]. Recurrent epileptiform-type neuronal discharges have been observed in neural networks of surviving neurons, and transient depolarizations occur in the ischaemic penumbra after experimental occlusion of the middle cerebral artery [29].
The mechanism of seizure initiation by haemorrhage is less clearly clarified. Blood products in the parenchyma and derivatives of heme and iron metabolism have been hypothesized to favour a focal cerebral irritation. Remarkably, the haemorrhagic transformation of an ischaemic brain infarct is associated with a meaningful higher risk of acute symptomatic seizures than ischemic stroke alone, providing further support to the role of blood extravasation in the development of abnormal epileptiform activity [26].
Cortical involvement is a well-characterized risk factor for early seizures in both ischaemic [20] and haemorrhagic stroke [30, 31]. Accordingly, the stratification by stroke subtype and location is essential in assessing an individual's risk of acute symptomatic seizures [29].
Severity of neurological deficit, lesion size, and younger age (<65 years) have also been associated with early seizures in some studies. Metabolic disturbances, such as high blood glucose or low sodium levels, can precipitate seizures. Alcohol use, previous stroke, low Alberta Stroke Program Early CT Score (ASPECTS) and low admission blood pressure levels have been suggested to increase the risk of acute symptomatic seizures after stroke [32].
Recently, a large retrospective analysis has identified non-neurological infections and a low premorbid functional level as shared risk factors for acute symptomatic seizures in haemorrhagic and ischaemic stroke [22, 33].
The concept that cardiogenic emboli are more likely to cause acute seizures is still controversial. The definition of cardioembolic stroke and the diagnostic work-up to assess cardiac embolic sources has differed widely across studies and the frequent association of cardiac embolism with cortical involvement represents a potential confounder [29].
Acute stroke therapies including intravenous thrombolysis and mechanical thrombectomy may potentially affect the risk of seizures, either increasing or decreasing the risk. In experimental models, recombinant tissue plasminogen activator has either been shown to have both neurotoxic and proconvulsant effects, such as loss of GABAergic inhibitory interneurons, upregulation of matrix metalloproteinases, enhancement of blood-brain barrier damage, and excessive production of nitric oxide, or neuroprotective and anticonvulsant properties, such as stimulation of brain-derived neurotrophic factor, inhibition of apoptosis and stabilization of cellular energy supply [34]. Sudden changes in cerebral perfusion and re-establishment of brain circulation may trigger an inflammatory cascade contributing to the development of the reperfusion syndrome and subsequent seizures. On the other hand, successful recanalization may limit the extent of the primary brain lesion [35]. A prospective study of 516 patients with ischaemic stroke did not find a significant difference in the incidence of acute symptomatic seizures in those patients who received reperfusion therapy (intravenous recombinant tissue plasminogen activator and endovascular thrombectomy) compared to those who did not [36]. Conversely, in a case-control study undertaken at a single stroke centre, intravenous thrombolysis was independently associated with the occurrence of acute symptomatic post-stroke seizures [37]. So far, there are not enough data to draw definitive conclusions, and the actual impact of acute stroke treatments on the risk of seizures remains unclear [34].
Few studies have explored EEG abnormalities as predictors of early post-stroke seizures. Lateralized periodic discharges (LPDs) and frontal intermittent rhythmic delta activities (FIRDAs) were found in 25% and diffuse slowing in about 30% of patients with acute symptomatic seizures. These EEG abnormalities were significantly more frequent in stroke patients with acute symptomatic seizures than in those without acute or subsequent late-onset seizures [38]. LPDs are considered as an unstable neurobiological process with an ictal-interictal continuum leading to overt seizures when acute metabolic derangements coexist [39]. In experimental models, LPDs appear over penumbral regions following middle cerebral artery occlusion, compared with intermittent rhythmic delta activity that occurs in the contralateral hemisphere with frontoparietal dominance [40].
Recently, the correlation between blood biomarkers and acute symptomatic seizures after stroke has been explored. Among a panel of 14 biomarkers collected within six hours of admission and before administration of any treatments, higher neural cell adhesion molecule (NCAM) and lower tumour necrosis factor receptor 1 (TNF-R1) levels were found to be independent predictors of early-onset seizures. NCAM plays a role in cellular adhesion and synaptic plasticity. TNF-R1 is widely involved in inflammatory pathways, and lower levels may be related to binding to TNF-α, a pro-inflammatory cytokine with proconvulsive effects [41]. Despite the explorative nature of this research, the combined use of clinical and electroencephalographic variables with serum biomarkers could be an interesting strategy for identifying patients at higher risk of early seizures.
In both ischaemic and haemorrhagic stroke, acute symptomatic seizures are predictive factors for the development of subsequent epilepsy [9, 42]. However, antiseizure medication (ASM) started at the time of acute symptomatic seizures has not been shown to reduce the risk of later epilepsy and is not recommended [43].
Cerebral venous thrombosis (CVT) is associated with a particularly high risk of acute symptomatic seizures which are the presenting symptom in 40% and may also occur after the diagnosis of CVT during the acute phase [44]. Risk factors for seizures associated with CVT include supratentorial, particularly haemorrhagic, brain lesions, neurological motor and sensory deficits and thrombosis of the superior sagittal sinus and cortical veins [45]. Seizures have not been reported to be an independent predictor of outcome. In one study including 153 patients with CVT, no differences were found in the mortality rate and functional status at six months after presentation between patients with acute symptomatic seizures and patients with no seizures [46]. Acute symptomatic seizures were found to be a predictor for the development of late seizures and epilepsy in patients with CVT in a recent, large retrospective study [47].
Infectious causes
Infectious diseases of the CNS represent a significant aetiology of acute symptomatic seizures. Acute symptomatic seizures occur in around 20% of patients with CNS infections [48, 49].
CNS infections cause acute symptomatic seizures in both adults and children, but the spectrum of CNS infections is different in both groups (e.g. bacterial meningitis is more common in children). Most studies have examined acute symptomatic seizures in association with CNS infections either in the adult or in the paediatric population. A review of the literature of acute symptomatic seizures in CNS infections needs also to consider that the prevalence and spectrum of CNS infections in both children and adults differ between geographical regions.
The risk of acute symptomatic seizures seems to be highest for viral encephalitis. Among a group of 147 adult patients with different CNS infections, patients with viral encephalitis were 14 times more likely to develop seizures than patients with other CNS infections such as bacterial meningitis [49]. In a prospective study of 148 patients with viral encephalitis, acute symptomatic seizures occurred in 42.6% [50]. Seizures in this study were both focal and generalised, and SE occurred in a quarter of patients. Of note, SE in encephalitis seems to be more refractory than with other aetiologies [51]. The reported seizure rates in encephalitis are probably underestimated as more subtle seizures and non-convulsive status epilepticus (NCSE) might be easily overlooked in a patient with symptoms of altered mental status if continuous EEG monitoring is not deployed [52]. A depressed level of consciousness, cortical lesions on brain MRI and younger age have all been reported as predictors of seizures in viral encephalitis [50]. Seizures in encephalitis are associated with a poor outcome [50] and increased mortality [53]. Encephalitis caused by herpes simplex virus (HSV) type 1 is the most common cause of sporadic encephalitis and the most strongly associated with acute symptomatic seizures in up to 60% in the acute stage [54]. The high seizure frequency seen in HSV encephalitis might be explained by the predilection of HSV to infect the epileptogenic mesial temporal brain structures including the hippocampus [55]. In vitro HSV infection of rat hippocampal cells has been shown to alter the excitability of hippocampal cells and to directly induce epileptiform activity [56]. High rates of acute symptomatic seizures are also reported for Japanese encephalitis, the most common form of an epidemic viral encephalitis. In a prospective study of 144 patients with Japanese encephalitis, acute symptomatic seizures occurred in 41% [53]. The presence of seizures in these patients was associated with raised intracranial pressure and clinical signs of herniation, with death four times more common than in those without seizures. Acute seizures in viral encephalitis may develop due to virus-induced neuronal cell death per se, however, the inflammatory response to the viral infection is probably more crucial. Cytokines, such as TNF-α, IL-1β and IL-6, have been shown to alter synaptic transmission, thereby leading to enhanced neuronal excitability [54]. The pathophysiology of acute symptomatic seizures in viral encephalitis is believed to be distinct from that causing late seizures and epilepsy, but both remain poorly understood. However, early seizures in encephalitis are associated with a higher risk of late seizures and epilepsy. In a study of 714 patients with encephalitis, the 20-year risk of developing unprovoked seizures was 22% for patients with viral encephalitis plus acute symptomatic seizures and 10% for those without seizures [48].
Bacterial meningitis is another frequent infectious cause of acute symptomatic seizures in both children and adults. A combination of the purulent inflammatory response and the direct effect of bacterial toxins is thought to trigger cortical inflammation that results in acute seizures [57]. In a prospective study of 185 children with bacterial meningitis, 31% developed acute symptomatic seizures [58]. Age below two years, infection with streptococcus pneumoniae, altered mental status, and a CSF leukocyte count below 1,000 cells have been determined as independent predictors of acute symptomatic seizures in children with bacterial meningitis [59]. Children with bacterial meningitis and acute symptomatic seizures appear to have a higher mortality rate than those without seizures [59]. In adults with bacterial meningitis, acute symptomatic seizures have been reported in 17-27% of cases [60, 61]. The majority of seizures occurs within the first 24 hours of presentation and seizures are more often generalised than focal [62]. Risk factors for acute symptomatic seizures in adults with bacterial meningitis are a decreased level of consciousness on admission, a positive CSF culture for streptococcus pneumoniae, a CSF cell count lower than 1,000 cells, elevated CSF protein and focal abnormalities on brain imaging [60]. Several studies have reported higher mortality rates for patients with acute seizures from bacterial meningitis [60, 61]. For both viral encephalitis and bacterial meningitis, the worse outcome reported in patients with acute symptomatic seizures than in those without seizures probably indicates a greater severity of the underlying disease in those patients with acute symptomatic seizures.
Acute symptomatic seizures are frequently encountered in other infectious CNS conditions. Up to 25% of patients with a brain abscess present with seizures [63]. Acute symptomatic seizures occur in more than 60% of children with cerebral malaria [64]. Seizures are the most frequent symptom in neurocysticercosis, occurring in 60-90% of patients [65]. In particular, seizures that occur in the presence of a degenerative cyst from neurocysticercosis with oedema on brain imaging should be considered as acute symptomatic seizures [5]. After resolution of the acute inflammatory lesion, recurrence of seizures is not the case for the majority of patients [66]. Seizures that occur after the resolution of the inflammatory lesion and in the presence of a calcified cyst should be considered as unprovoked seizures and warrant sustained antiepileptic treatment [67]. Acute symptomatic seizures have been reported in up to 20% of HIV patients and in about 4% at the time of presentation [68]. Opportunistic infections, such as cerebral toxoplasmosis, cryptococcal meningitis or progressive multifocal leukoencephalopathy, are the most frequent causes of seizures in HIV patients and the majority of patients develop epilepsy [69, 70].
Autoimmune and inflammatory causes
A very heterogeneous group of primarily immune-mediated disorders are variably associated with acute symptomatic seizures and include those that affect solely the CNS such as autoimmune encephalitis or multiple sclerosis and those that typically cause systemic symptoms with occasional CNS involvement such as systemic lupus erythematosus.
There is a very strong association with acute symptomatic seizures and non-infectious, immune-mediated forms of encephalitis [71]. Two groups of these disorders are distinguished by their different pathophysiological and clinical characteristics: cancer-triggered inflammatory responses against the nervous system, the so-called paraneoplastic syndromes [72] and antibody-mediated autoimmune encephalitis syndromes. In paraneoplastic syndromes, the immunological mechanism is mediated by cytotoxic T-cells, accompanied by the presence of non-pathogenic antibodies against intracellular antigens. The risk of acute symptomatic seizures depends on the brain regions involved and is particularly high if the limbic system is involved [72]. The more chronic and treatment-refractory inflammatory disease process in these disorders favours an enduring predisposition for the generation of seizures so that many patients (up to 60%) develop epilepsy [72]. In the antibody-mediated autoimmune encephalitis syndromes, an immune response, which is usually of an unknown cause although may be triggered by a tumour or a preceding viral encephalitis, generates antibodies directed against neuronal surface antigens [73]. The disease is the direct result of the pathogenic interaction of antibodies with their neuronal surface antigens, and the clinical manifestations vary according to the specific antigen. Seizures are often the leading symptom of antibody-mediated autoimmune encephalitis and develop in 33% to 100% of all patients, depending on the target antigen [72]. Autoimmune encephalitis is the most commonly identified cause of new-onset refractory status epilepticus (NORSE) [74]. The antigens most strongly associated with seizures are N-methyl-D-aspartate receptor (NMDA) receptor, gamma-aminobutyric acid (GABA) B receptor, GABA A receptor and leucine-rich, glioma inactivated 1 (LGI1, part of the voltage-gated potassium channel complex). The majority of patients with antibody-mediated autoimmune encephalitis have a favourable outcome when treated appropriately with immunotherapy [73]. Seizures usually vanish when the encephalitis resolves. Thus, seizures in antibody-mediated autoimmune encephalitis, even if they occur over a prolonged period of time, are acute symptomatic seizures in a true sense, as elimination of the precipitating antibody typically stops the seizures. With the exception of LGI1 and GABA A receptor encephalitis, the risk for recurrent seizures after the acute inflammatory phase of antibody-mediated autoimmune encephalitis is low (e.g. < 5% in NMDA receptor encephalitis) [72].
Seizures are an infrequent but long and well recognised feature of multiple sclerosis [75]. Around 2-5% of all patients with multiple sclerosis develop one or more epileptic seizures during their lives [76]. A seizure that occurs as the first presenting symptom of multiple sclerosis as well as seizures that occur only during a relapse should be considered as acute symptomatic seizures [5]. A seizure will be determined as part of a relapse if it is accompanied by the occurrence or worsening of other neurological symptoms or signs of multiple sclerosis or if there is a typical active demyelinating lesion adjacent to the cerebral cortex detected on brain imaging. If seizures recur and are not related to a relapse, epilepsy should be diagnosed. The relative frequencies of acute symptomatic seizures and unprovoked seizures in multiple sclerosis are unclear. In a cohort of 268 multiple sclerosis patients, 20 patients with epileptic seizures were identified of whom four had seizures as the presenting manifestation, eight had seizures solely during a relapse and 12 had unprovoked seizures [77]. Seizures that occur only during a relapse will not require long-term antiseizure treatment.
Epileptic seizures occur in approximately 15% of patients with systemic lupus erythematosus (SLE) [78]. A study of 60 SLE patients with epileptic seizures reported that 88% of patients had acute symptomatic seizures and only 12% had recurrent seizures [79]. Seizures occur mainly at the disease onset or during disease flares or may be caused by a stroke in the setting of a prothrombotic state or vasculitis, CNS infection, hypertension/PRES, renal insufficiency or probably by direct CNS effects of antiphospholipid antibodies [78].
Other systemic autoimmune disorders associated with seizures include Sjögren's syndrome, granulomatosis with polyangiitis, sarcoidosis, celiac disease, Crohn's disease and Behcet's disease. [78]. There are many causes of seizures in these disorders including direct immunological effects on the brain via cytokines, immune complexes and autoantibodies as well as vascular disease, infections, metabolic disorders and adverse effects of immunosuppressive therapies.
Steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT), also known as Hashimoto encephalopathy (HE), refers to a vaguely defined syndrome of an acute/subacute encephalopathy that is associated with the presence of elevated autoantibodies against thyroid peroxidase (TPO) and/or thyroglobulin (TG) in the absence of other causes [80]. An autoimmune aetiology is assumed because some patients respond well to corticosteroid therapy within days to weeks and because of the presence of thyroid autoantibodies. There is no clear evidence that thyroid autoantibodies are pathogenic in SREAT. As these antibodies have a high prevalence in the general population, they might just represent an completely unrelated finding [81] or non-specific evidence of a predisposition to autoimmunity [81]. Acute symptomatic seizures of varying types have been reported in 70-80% of SREAT patients and SE in 10-20% [76]. In a recent study of 24 patients with suspected SREAT, who had no evidence for other neurological disorders including negative CSF/serum antineuronal antibodies, four distinct clinical subtypes were recognised: patients with solely psychiatric symptoms (29%) and patients with signs of encephalopathy (29%) who had no seizures in contrast to patients with limbic encephalitis and seizures (17%) and patients presenting with NORSE (25%) [82]. Steroid-responsiveness was seen in 32% of all patients but there were no features that allowed predicting steroid-responsiveness among patients. It seems likely that SREAT is a group of heterogeneous disorders with different underlying causes that are not yet clear [82]. The presence of thyroid autoantibodies in a patient with new-onset seizures and no other obvious cause should not lead to an immediate diagnosis of SREAT and does not predict a response to immunotherapy for such a patient.
Brain trauma
Acute symptomatic seizures (within one week) that are clinically evident after moderate-severe traumatic brain injury (TBI) occur in 2-15% of patients, with most studies showing an incidence of around 3-5%; about half occur in the first 24 hours [83-85]. Higher rates of seizures are seen when continuous EEG monitoring is performed (see next section). Antiseizure medications (ASMs) given prophylactically after a head injury can decrease the rate of early seizures (first week), but have no effect on later epilepsy [83, 85]. The most carefully studied ASM have been phenytoin and levetiracetam. Patients with apparent seizures at the moment of impact, so-called “impact fits” or “concussive convulsions”, are at low risk of further seizures, probably lower than those with other early seizures during the first week of injury [86].
Risk factors for acute seizures after TBI include more severe injury, need for neurosurgical intervention, depressed skull fracture, younger age (much higher in young children than adults), penetrating injury, and any type of intracranial haemorrhage. As with other conditions discussed above, acute symptomatic seizures after TBI are a major risk factor for development of epilepsy, with an odds ratio of 5 in a meta-analysis of risk factors for the development of post-traumatic epilepsy [87]. One study found that acute epileptiform abnormalities on EEG were an independent risk factor for later epilepsy with on odds ratio of 3.2 after controlling for severity of TBI [88]. In animal models, acute high-frequency oscillations appear to be a biomarker of early post-traumatic epileptogenesis [89]; human studies including invasive EEG are underway to help address this and other potential biomarkers [90].
Acute seizures in the critically ill/ICU
In the past decade, continuous EEG monitoring (cEEG) in the critically ill has become standard of care in many centres [91]. This is due to the recognition that the majority of electrographic seizures detected via cEEG (about 75% overall) have no detectable clinical signs at the bedside and would be missed without recording EEG [92-94]. Of all critically ill patients undergoing cEEG, about 15-20% have seizures, and many more have highly epileptiform patterns such as periodic discharges [93-95]. The rate of seizures is higher for many subgroups such as postanoxic coma, brain tumours, severe TBI with intracranial haemorrhage, non-traumatic lobar intracerebral haemorrhage, central nervous system infections, and after convulsive SE [93, 95, 96]. Other risk factors for developing acute electrographic seizures include prior clinical seizures (recent or remote), coma, acute supratentorial brain injury, and sepsis. A one-hour EEG will detect seizures in about half of these patients, and a 24-hour study will detect the first seizure in about 90% [92, 93, 95].
Prospective outcome studies have found that non-convulsive seizures, especially if prolonged or frequent (occupying >20% of the recording), are independently associated with worse outcomes, including hippocampal atrophy, overall function, cognition, and development of later epilepsy [97-99]. Studies in both children and adults and with different types of acute brain injuries have found a “dose-response” relationship to these adverse outcomes (higher seizure prevalence = worse outcome) [99, 100].
Although many have referred to a “silent period” between acute injury (with or without acute symptomatic seizures) and later epilepsy, EEG recordings in both animals and humans have cast doubt on this period being truly silent; perhaps a better term would be “clinically silent period” followed by “overt seizures” [101]. The acute EEG findings may be a biomarker of ongoing epileptogenesis. As mentioned above, epileptiform activity in the first five days after TBI are an independent predictor of later epilepsy [88]. In one study of patients with acute brain injury, 31% of patients with lateralized periodic discharges or electrographic seizures went on to develop epilepsy, compared to 4% in a matched control group without epileptiform EEG findings [102]. Similarly, a study of LPDs alone found that more than one third of patients with LPDs developed new-onset epilepsy, which was seven times as likely to occur than in matched controls without highly epileptiform EEG patterns [103].
It is yet to be determined whether acute intervention to decrease non-convulsive seizures or periodic discharges can lower the rate of subsequent epilepsy or other adverse effects.
Metabolic causes
Acute or severe electrolyte disturbances frequently cause seizures. Imbalances of electrolyte levels can alter the ion gradients between extra- and intracellular space and thereby cause changes in neuronal discharge that can result in altered neuronal excitability and synchronisation [104]. Seizures are most often associated with hyponatraemia and less frequently with hypernatraemia, hypocalcaemia and hypomagnesia. Seizure semiology is usually generalised tonic-clonic. The risk of seizures increases with the severity of the electrolyte imbalance and minor changes in electrolyte levels are not regarded as a sufficient cause for a seizure. Cut-off levels of electrolytes that support a probable causal relationship for a seizure have been proposed by the ILAE (table 2) [5]. Possibly the acuteness of change is more important than the absolute electrolyte level [10]. When electrolyte levels are chronically low, it is often difficult to convincingly establish a causal relationship with a seizure except when the derangement is extreme, for example if the serum sodium is <110 mmol/L.
Seizures can rarely occur in severe hypogylcaemia though the predominant neurological symptom is usually coma. In a retrospective study of 388 patients who had been admitted to hospital for evaluation of hypoglycaemia (blood glucose <60 mg/dL) only one generalised tonic-clonic seizure (blood glucose level <36 mg/dL) and two focal seizures (blood glucose level: 36 mg/dL and 59 mg/dL) were reported [105]. Hyperglycaemia, particularly non-ketotic hyperglycaemia, is known to cause acute symptomatic seizures as well, especially focal motor seizures [106].
Renal failure may cause seizures by electrolyte disturbance, on its own (serum creatinine > 10 mg/dL), or in the context of dialysis disequilibrium syndrome. Liver failure is an infrequent cause of seizures [107] except as a complication of liver transplantation. In a retrospective study of 146 patients who underwent liver transplantation, 15.7% of the patients had a seizure within one week after the procedure [108].
As metabolic derangements such as electrolyte disturbances usually do not affect the structural integrity of the brain, seizure recurrence is not expected once the causative abnormality has been corrected and metabolic homeostasis has been restored. Treatment is focused on the correction of the underlying pathology but ASMs are often used concomitantly in the acute phase.
Medications, intoxications and alcohol-related seizures
Medication-related seizures can result both from the ingestion or withdrawal of certain medications. In a retrospective study of 276 patients with new-onset seizures, 6.1% of seizures were found to be drug-related [109]. Cocaine intoxication, benzodiazepine withdrawal and bupropion were the three leading causes of drug-related seizures in this study. The most common medications associated with seizures are antidepressants (bupropion, citalopram, venlafaxine, trimipramine, amitriptyline, maprotiline), antipsychotics (clozapine, chlorpromazine, quetiapine), antihistamines (diphenhydramine) and analgesics (tramadol and mefenamic acid) [110-113]. However, the overall risk of seizures with these medications, especially when compared to their widespread use, seems low and should not prevent their use when needed. It has been suggested that some intravenous antibiotics carry an increased risk of acute symptomatic seizures including fourth-generation cephalosporins (especially cefepime [114]), carbapenems (especially imipenem [115]) and ciprofloxacin, predominantly when given at high doses and in patients with renal insufficiency, brain lesions or pre-existing epilepsy [116].
Seizures related to medication withdrawal occur most commonly with benzodiazepines and barbiturates. The use of propofol has been associated with seizure-like phenomena (SLP) in several case reports [117]. The cause of SLP with propofol has been debated and several authors have argued for their non-epileptic nature [118]. Cocaine and amphetamines are the recreational drugs that are by far most strongly associated with seizures [119]. Seizures can be potentially caused by certain hallucinogens while seizures are unlikely to be related to heroin or marijuana abuse [5].
Alcohol-related seizures are a very frequent cause of acute symptomatic seizures with some studies suggesting that up to one third of all seizure-related hospital admissions are alcohol-related [120]. Alcohol withdrawal is most often the cause of seizures, however, seizures can occur during severe alcohol intoxication as well. An alcohol withdrawal seizure is suspected in a patient with a history of chronic alcohol abuse and recently reduced alcohol consumption who presents 7-48 hours after the last alcoholic drink with a generalised tonic-clonic seizure along with typical symptoms of withdrawal such as tremor, sweating and tachycardia. Alcohol withdrawal seizures are thought to arise from a hyperexcitable brain state from changes in NMDA receptor and GABA A receptor signalling that develop during chronic alcohol abuse [121]. Caution is warranted when assigning a seizure as alcohol-related; in a retrospective study of 140 patients who presented with what was initially thought to be an alcohol-related seizure, alternative causes other than alcohol were identified in 53.6% of patients and included head trauma, epilepsy, stroke and metabolic abnormalities [122]. European guidelines recommend brain imaging and EEG even in obvious cases of a first alcohol-related seizure [120]. However, in many countries, EEG will not be part of the evaluation of a first alcohol-related seizure. In clinical practice, EEG is often not feasible, for instance when patients are lost to follow-up.
Other causes
Posterior reversible encephalopathy syndrome
In posterior reversible encephalopathy syndrome (PRES), reversible vasogenic brain oedema develops in the context of either severe hypertension/blood pressure fluctuations, renal failure, drug toxicity or autoimmune disorders that leads to acute neurological manifestations such as seizures, encephalopathy, headache and visual disturbances [123]. Endothelial injury due to either severe hypertension with loss of cerebral autoregulation or due to direct effects of cytokines results in the breakdown of the blood brain barrier, cerebral oedema and subsequently seizures. Brain imaging shows subcortical vasogenic oedema with a predilection for the parietooccipital regions in most patients. Acute symptomatic seizures (usually focal to bilateral tonic-clonic) occur in 60-75% of patients, often as the presenting symptom [124, 125]. These seizures appear to originate in the parietooccipital regions, correlating with the location of the typical abnormalities observed on cerebral MRI [126]. SE, especially NCSE has been reported in 5-15% of patients with PRES [123] with a posterior seizure focus on EEG [127]. In a study of 37 critically ill patients with PRES undergoing cEEG monitoring, non-convulsive seizures or periodic discharges were found in 62% of patients on cEEG monitoring and 16% of patients fulfilled EEG criteria for NCSE [128]. Non-convulsive seizures and periodic discharges were mainly found over the posterior region, especially in patients who had cortical diffusion-restricted lesions on cerebral MRI and were associated with a worse outcome. The clinical and imaging manifestations of PRES are usually reversible and around 75-90% of patients make a full recovery within one or two weeks [123]. The generally favourable prognosis goes along with a low risk of recurrent unprovoked seizures. In a cohort of 127 PRES patients with a median follow-up of 3.2 years, only two had a single unprovoked seizure and one developed epilepsy [129].
Eclampsia
Eclampsia is defined by the new onset of acute symptomatic epileptic seizures in a woman during pregnancy or after delivery who has signs and symptoms of preeclampsia such as hypertension, proteinuria and oedema [130]. Eclampsia is a rare complication of pregnancy with 1.5-10 cases per 10,000 deliveries and carries a significant risk for severe maternal morbidity and mortality [131]. Approximately a half of the seizures occur antepartum but seizures also occur relatively frequently during delivery and within the first days postpartum [132]. Seizures are focal to bilateral tonic-clonic, are often preceded by headache and visual disturbances and will usually develop in patients who have already exhibited signs of preeclampsia. However, seizures may develop in patients before the hypertension and proteinuria [133]. The pathophysiological process of eclampsia shares common pathways with PRES. Cerebral MRI demonstrates T2 hyperintense white matter lesions with a parietooccipital predilection similar to those seen in PRES [134]. Management includes the administration of magnesium sulphate, which reduces the risk of recurrent seizures significantly more effectively than diazepam [135] and phenytoin [136]. Magnesium sulphate is also effective for primary seizure prevention in patients with preeclampsia [137]. Women with eclampsia have a higher relative risk of developing epilepsy than women with uncomplicated pregnancies, even though the absolute risk of developing epilepsy after eclampsia is very small [138].
Cerebral anoxia
Anoxia due to cardiopulmonary arrest often causes cerebral damage that involves the thalamus, hippocampus and cortical pyramidal cells, and thus can lead to seizures [139]. Clinically overt seizures occur in one third of comatose patients with anoxic encephalopathy [140]. The majority develop myoclonus, either cortical and/or subcortical, whereas generalised tonic-clonic seizures only occur in 7% of all patients with anoxic encephalopathy. Myoclonus will progress to myoclonic SE in a minority of patients. NCSE has been reported in up to one third of comatose patients with anoxic encephalopathy [141]. Myoclonus/myoclonic SE and NCSE have been long viewed as predictors of poor neurological outcome, however more recent literature suggests that good neurological outcome is still possible [139]. Convulsive seizures in patients with anoxic encephalopathy should be treated with ASM, as for any other acute disorder. Benzodiazepines, valproic acid and levetiracetam may be most effective for myoclonic seizures [142]. There is no consensus on how aggressively NCSE should be treated in patients with anoxic encephalopathy due to a lack of evidence [143]. However, increasing reports of patients with post-anoxic refractory NCSE and good neurological outcome seem to justify more aggressive and prolonged treatment with ASMs and sedatives [144].
Management
Successful treatment of acute symptomatic seizures first requires that a seizure is recognised as acute symptomatic and that immediate diagnostic measures are undertaken to identify the underlying pathology. Information from the patient's history and the findings on the physical and neurological examination will lead to the hypothesis that a seizure might have an acute symptomatic cause. This should then be further evaluated with a routine laboratory investigation and brain imaging, as well as further investigations such as EEG or a lumbar puncture if indicated. Once a diagnosis is made, causal treatment of the underlying disease should be established as soon as possible (e.g. i.v. thrombolysis and mechanical thrombectomy for ischaemic stroke, acyclovir for herpes encephalitis, anticoagulation for cerebral venous thrombosis, etc.).
Patients with acute symptomatic seizures should be treated with ASMs during the acute phase of the underlying disease or as long as the precipitating factor or cause is present. Patients with acute symptomatic seizures can develop further acute symptomatic seizures soon after the first event [145]. The aim of treatment with ASMs is to prevent further acute symptomatic seizures. However, data on how long to treat patients with acute symptomatic seizures with ASMs is largely lacking. The duration of treatment with ASMs needs probably to be individualised for every patient and will depend on factors such as the underlying aetiology (e.g. fully resolved hyponatraemia versus an ongoing autoimmune encephalitis), the risk of further seizures, the severity of the acute symptomatic seizure (e.g. SE, seizure-related injuries), the overall clinical situation (e.g. Is the patient awake and doing well or is the patient still critically ill/comatose in intensive care? Are there severe comorbidities?), and patient preference. For patients with an acute structural brain insult, one can extrapolate from the TBI literature and conclude that one to two weeks of treatment with ASMs will lower the rate of early seizures, but longer treatment is unlikely to be effective and will unnecessarily expose patients to potential adverse effects. This is logical and likely the most evidence-based approach currently feasible. However, in many clinical situations, treatment with ASMs for longer than two weeks might be necessary and clinical judgement that considers individual patient factors is required in the decision-making process. Long-term antiepileptic treatment is generally not indicated, based on the significantly lower recurrence risk of acute symptomatic seizures than unprovoked seizures, as already discussed above [8]. However, in our experience, many patients are left on ASM treatment for relatively long periods, often indefinitely. It seems reasonable to perform an EEG two to three months after hospital discharge for anyone remaining on ASMs due to acute symptomatic seizures or highly epileptiform EEGs. If the EEG does not show epileptiform discharges, ASMs are tapered off. Slowly tapering the ASM might reduce the risk of a seizure occurring from the discontinuation of the ASM itself. The EEG is sometimes repeated once ASMs have been discontinued, especially if the patient had been treated with benzodiazepines or levetiracetam which might “hide” spike discharges. This approach is pragmatic but not evidence-based. If possible, the patient should be involved in the decision-making process regarding the duration of ASM treatment. This includes explaining to the patient that his or her risk of seizure recurrence after an acute symptomatic seizure is generally low, but that there might be a substantial risk of further seizures in some situations.
Case 1
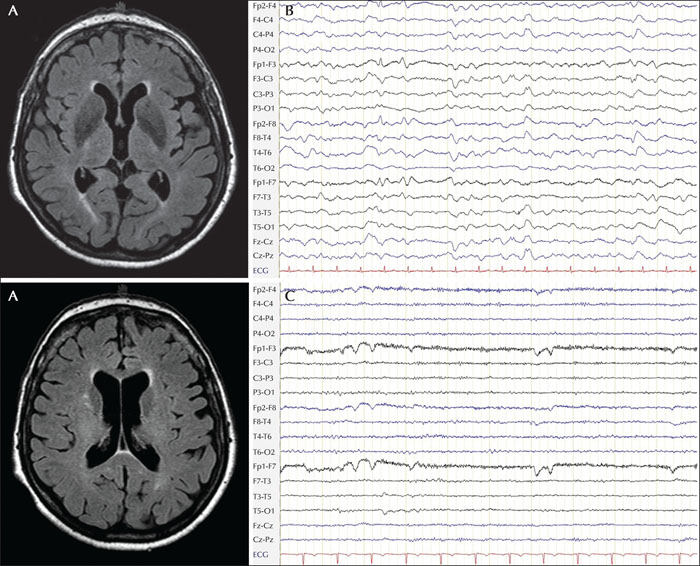
An 84-year-old woman was found unconscious on the floor of her flat and was brought to hospital. The patient's past medical history included hypertension treated with ramipril and hydrochlorothiazide, chronic kidney disease and an episode of hyponatraemia which had previously required admission to the internal medicine department. There was no history of seizures or other neurological diseases. She was living independently without any signs of cognitive decline. In the neurology emergency room, she developed a generalized tonic-clonic seizure and remained unresponsive thereafter. Brain MRI revealed moderate subcortical vascular changes and moderate generalised atrophy, but did not show an ischaemic stroke, bleeding or intracranial vessel occlusion (figure 3A). EEG showed diffuse slowing, compatible with encephalopathy without any epileptiform changes (figure 3B). Serum sodium was 108 mmol/L. The patient was admitted to the intensive care unit and made a full recovery after careful electrolyte correction. Treatment with levetiracetam, which had been started on admission, was stopped half a year later at a follow-up visit when an EEG examination was normal (figure 3C). At a follow-up visit four years later, the patient did not report any further seizures.
Case 2
A 48-year-old woman was transferred to the neurology emergency department for urgent evaluation after an episode of unresponsiveness. The patient had been treated for hypertension and hyperlipidaemia in the past and had been smoking 15 cigarettes per day for the last 20 years. She reported that she had felt numbness in her left arm for two minutes but could not remember what had happened thereafter. The patient's daughter had noticed her suddenly staring motionless without any response for one minute. Afterwards, the patient did respond again to her daughter but appeared confused. Neurological examination was remarkable for a subtle paresis of the left arm and a mild facial palsy. Brain MRI showed multiple acute cortical ischaemic lesions on diffusion-weighted imaging (DWI) in the territory of the right middle cerebral artery, particularly in the posterior third (figure 4A), and a high-grade stenosis of the right internal carotid artery at the level of the carotid bifurcation was detected on MR-angiography (figure 4C). Because most ischaemic lesions seen on DWI were also already visible on T2-weighted sequences, intravenous thrombolysis was not administered (figure 4B). EEG showed focal slowing over the right posterior hemisphere, but no epileptiform discharges were visible (figure 4D). The patient was treated with carotid artery stenting. Levetiracetam was administered for two weeks and subsequently tapered before she was discharged home. On follow-up, only a mild paresis of the left arm was noted and no further episodes, suspicious for seizures, have occurred.Key points
Supplementary material
Summary slides accompanying the manuscript are available at www.epilepticdisorders.com.
Disclosures
Lawrence Hirsch has received consultation fees for advising from Accure, Aquestive, Ceribell, Marinus, Medtronic, Neuropace and UCB; Royalties from Wolters-Kluwer for authoring chapters for UpToDate-Neurology, and from Wiley for co-authoring the book “Atlas of EEG in Critical Care”, by Hirsch and Brenner; and honoraria for speaking from Neuropace and Natus.
Richard Chin reports personal fees from GWPharma, Eisai, Zogenix; his institution received grants from GWPharma, Eisai, Zogenix, the Norwegian Research Council, the Wellcome Trust, the Medical Research Council, Epilepsy Research UK, Epilepsy Action, the Muir Maxwell Trust, and the RS McDonald Trust outside the submitted work.
Simona Lattanzi has received speaker's or consultancy fees from Eisai, GW Pharmaceuticals, and UCB Pharma and has served on advisory boards for Angelini, Arvelle Therapeutics, BIAL, and GW Pharmaceuticals.
Eugen Trinka reports personal fees from EVER Pharma, Marinus, Arvelle, Argenix, Medtronic, Bial-Portela & Cª, NewBridge, GL Pharma, GlaxoSmithKline, Boehringer Ingelheim, LivaNova, Eisai, UCB, Biogen, Genzyme Sanofi, and Actavis; his institution received grants from Biogen, UCB Pharma, Eisai, Red Bull, Merck, Bayer, the European Union, FWF Osterreichischer Fond zur Wissenschaftsforderung, Bundesministerium für Wissenschaft und Forschung, and Jubiläumsfond der Österreichischen Nationalbank outside the submitted work.
Matthias Mauritz, Peter Camfield and Raffaele Nardone have no conflicts of interest to declare.