Virologie
MENUViral persistence of mammalian reovirus in cell culture: a model of virus-cell coevolution Volume 23, issue 5, Septembre-Octobre 2019





General principles
Cytolytic viruses, such as mammalian reovirus (hereafter referred to as “reovirus”) can sometimes establish persistent nonlytic infection in tissue culture. Although this is outside the scope of the present review, the importance of “revisiting” the concept of “cytolytic” viruses was recently stressed [1]. Indeed, mammalian reovirus, although generally considered as cytolytic, can be released from certain cell types in the absence of cell lysis [2, 3].
The phenomenon of viral persistence, thus far from being only a laboratory curiosity, can constitute a powerful experimental system to decipher important steps in the viral replication cycle. The establishment of viral persistence can be accompanied by important changes in the properties of the virus itself, but also of its host cell, and thus constitutes a biologically significant model of virus-host coevolution.
Among mammalian viruses, reovirus has been extensively used as a model of viral replication and viral pathogenesis. More recently, a renewed interest in the study of this virus has been noticed since it is currently used in clinical trials for virotherapeutic approaches against various cancers [4, 5]. Reovirus, under his trade name Reolysin®, has in fact been granted the status of orphan drugs for various cancers by both the Food and Drug Administration (FDA) in the United States and the European Medicines Agency (EMA).
Not surprisingly, this virus, despite the fact that it is generally cytolytic, has been the subject of various studies concerning its ability to establish and maintain viral persistence, essentially in cell culture. Such studies had an important impact on our understanding of reovirus replication and host-cell interactions in the past and still have the potential to further contribute to this understanding. Furthermore, such approaches could help to develop practical applications for reovirus, by allowing the selection of new viral variants that are better adapted to infect and destroy cancer cells, among other things [6, 7].
Surprisingly, despite the fact that viral persistence has now been obtained from many different cell lines, few of the resulting adapted viruses had their genomes completely sequenced. Indeed, only the sequence of the Vero-cell-adapted virus obtained in our laboratory has been completely determined to date [8]. The progress in sequencing approaches will certainly allow to correct this situation in the near future. Furthermore, the advent of plasmid-based reverse genetics now allows to confirm the importance of the different mutations observed in the adapted viruses. These mutations can be separately introduced in a wild-type virus backbone, as done in some of our recent works [8, 9].
As discussed later, early events in the viral multiplication cycle, leading to virus entry, are central to the phenomenon of viral persistence (figure 1) [10-12]. Following the binding of the viral σ1 protein onto the cell surface, the viral particle will be internalized. The σ1 protein possesses a long helicoidal portion in its amino-terminal region, including a sequence at the end for anchoring the protein to the virion. The central region of the protein allows binding to sialic acids in virus strains that actually possess this property, including the Dearing strain of serotype 3, generally used in the studies of viral persistence. The protein ends by a globular region at the carboxyl-terminal end involved in the binding to the protein receptor mostly used for virus binding to epithelial cells, the junctional adhesion molecule (JAM) protein [10, 13]. The cleavage of outer capsid proteins, beginning by the σ3 outer capsid protein, first follows internalization of the viral particle. Cleavage of the outer capsid protein μ1 then allows to generate infectious particles referred to as “infectious subviral particles” (ISVPs) that can then cross the endosomal membrane toward the cytoplasm, allowing activation of the transcription from the inner viral capsid or “core.” Partial uncoating by lysosomal enzymes, to generate ISVPs, is a limiting factor in a number of cell types [14-18]. The ISVPs can also be generated by proteolytic cleavage outside the cells, allowing to bypass the steps of intracellular uncoating. It should be mentioned that the structure of σ1 and of the heterohexameric complex σ3-μ1 [19-21] has been determined and will be discussed later on (figure 3andfigure 4).
About a dozen different cell lines have been reported to become persistently infected by reovirus under certain conditions. However, this depends on the definition given to “persistent infection.” As an example, infection of the bat Tb1-Lu cell line results in a productive infection with minimal cell death [22]. Viral production appears to slowly decrease with time without actual changes in either cells or viruses. This should be rather called “nonlytic” or “chronic” infection, rather than persistent infection per se, as will be discussed later. In some cell types in which persistent infection was reported without actual cytopathic effect, such as in MDCK cells [23], it is probably a similar situation in which viral persistence with all its consequences is not necessarily established. Our own data were, in fact, quite different in this cell line (unpublished data). This raises the question of variations in the cell lines and viruses used between laboratories, thus further complicating comparisons. Furthermore, in many cases, viruses and cells were poorly characterized following the establishment of persistence. Another example is the report that persistent reovirus infection affects growth properties of BALB/c 3T3 murine fibroblasts [24]. In later work it has rather been demonstrated that reovirus preferentially infects transformed murine fibroblast cells while parental cells, such as untransformed BALBc cells, are quite resistant to the infection [25, 26]. This suggests that the cell stock used during the establishment of viral persistence may have been partly made of transformed cells, as occurs when cells are let to stay confluent for a certain period of time. Alternatively, contamination by another cell type remains a possibility.
In this review, persistent reovirus infection will be defined as an infection that is established after a period of cell lysis in cell types in which infection is cytolytic under usual conditions. The majority of the cells are then infected but constant reinfection is necessary to maintain the infection. We will see later on that this is accompanied by changes to both the virus and the cells involved.
We will now examine in greater details few cellular models in which reovirus persistent infection was studied more extensively.
The classical L929 cells model
Initially, most of the work on viral persistence was done using L929 cells, which are mouse fibroblasts most often used for reovirus growth. Viral persistence in this cell model has been reviewed in 1998 [27]; we will briefly summarize here the major aspects of this model and further discuss progress made since this last extensive review.
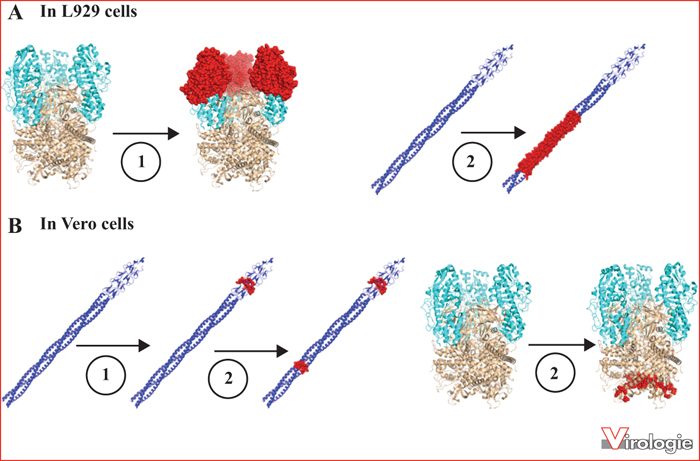
Upon reovirus infection, most L929 cells are readily killed and destroyed, sometimes in less than 24 hours at 37 °C, depending on multiplicity of infection that was used. However, it was observed that a small number of cells can nevertheless survive. These cells will slowly grow with frequent phases of “crisis” in which extensive cell death is observed. Nevertheless, following this relatively long period of persistence establishment, a cell population can be selected. These cells continuously produce and release infectious virus in the absence of cell lysis; the virus produced is being referred to as a PI virus (from Persistent Infection). The persistently infected cells can eliminate the virus by long-term exposure to a neutralizing antireovirus antiserum [28]. Interestingly, these so-called “cured” cells are quite resistant to the wild-type virus but easily killed upon infection by the corresponding PI virus. A schematic summary of the establishment of viral persistence on L929 cells is presented in figure 2A, while figure 2B schematizes the result of virus-cell coevolution. Further work has shown that cured cells have been selected to express reduced levels of mature lysosomal proteases known as cathepsins [29, 30]; these cells are therefore inefficient at removing the outer viral capsid proteins during viral uncoating. In parallel, PI viruses have evolved to be able to infect the cured cells. Studies using gene exchange (reassortment) have initially found that a major outer capsid protein, σ3, and the viral cell attachment protein, σ1, are respectively involved in establishment and maintenance of viral persistence [31, 32]. To date, the complete genome sequence of a PI virus from L929 cells has not been obtained, but the sequence of the genes encoding these two apparently critical proteins has been. Actually, this sequencing was not performed directly on PCR products, as is now most commonly used, but rather by cloning of cDNAs, always raising the possibility of sequencing a minor population that is not representative of the actual replicating virus. It will certainly be of interest to confirm these sequences on different populations of PI viruses. However, the study of amino acids substitutions on σ3, especially the Y354H substitution, has allowed to clearly establish that these substitutions render the virion more sensitive to cathepsins [33]. This allows uncoating in cured cells while the wild-type virus is blocked under such reduced proteases conditions. Accordingly, PI viruses are resistant to inhibitors of endosomal acidification (required for the formation of mature cathepsins) and, more directly, to lysosomal protease inhibitors such as E64 [33-37]. Virions of PI viruses are thus in a sense reminiscent of ISVPs. The X-ray crystal structure allows to localize the amino acid substitutions on the σ3 protein of different known PI viruses, as shown in figure 3. As discussed later on, substitutions located on the σ3 protein are also found on PI viruses obtained from other cell lines. The molecular structure is available for the homodimer of σ3, as well as the σ3-μ1 heterohexamer, the major constituent of the outer capsid, and was later refined by cryomicroscopy of the virion [19-21]. These different substitutions are all located on the small external lobe of the protein, and thus likely to affect the proteolytic removal of σ3 [10, 12, 38, 39]. These substitutions are located at the surface of the protein and away from the contact zone between the two copies of the molecule in its dimeric form. The substitutions are also away from the contact zone between σ3 and μ1 in the heterohexameric complex at the virion's surface.
One can wonder why wild-type viruses have not acquired this ability to more easily uncoat, that could be seen as an advantage. In fact, such a virus is more pathogenic and is being transmitted more efficiently in an animal model [40]. It is, however, possible that an increased pathogenicity is actually harmful for the virus on the long-term; alternatively, it is also possible that the transmission assay used does not exactly reflect the negative impact of decreased virion stability in the environment. Nevertheless, more detailed studies of reovirus strains in natural situations seem necessary to better understand if such mutations arise or not, and are maintained or not, under such conditions.
The exact role of the σ1 substitutions in these viruses remains elusive; it was suggested that they can affect the stability of the complex formed by three copies of the protein [41] but the precise impact was not further studied. The determination of the structure of the protein [42, 43] now allows us to locate more precisely the substitutions, confirming their localization in the helical region of the protein (figure 4) unambiguously allowing to affirm that they exert an effect on the stability of the protein [42]. It should also be mentioned that some of the mutations overlap the open reading frame for the small σ1s protein, encoded by the same S1 gene; it is thus difficult to affirm that the phenotype is really due to changes on σ1. The advent of reverse genetics could probably lead to a further understanding of the importance of these various substitutions. Interestingly, in two other different cell types, persistence is also accompanied by changes in σ1 (or σ1s). This aspect will be discussed in a subsequent section.
The Vero cell situation
In the last few years, our laboratory has characterized in detail a virus obtained following reovirus persistence in Vero cells. These cells were chosen since they apparently possess a limited ability to uncoat the virus [14], raising the hypothesis that adaptation could occur by a mechanism other than modification of uncoating. These cells also exhibit well-known defects in the antiviral interferon-induced, double-stranded-RNA-dependent, protein kinase (PKR) [44-47]. The genome sequence of the PI virus obtained named Vero-cell-adapted virus (VeroAV) was completely determined and revealed amino acids substitutions in four of the eleven translation products of the ten viral genes [8]. The use of the reverse genetics system then allowed us to demonstrate that the single amino acid substitution N198K in σ1 is responsible for adapting the virus to Vero cells by increasing its binding to sialic acids (figure 4) [9]. Interestingly, in this case, there was no change in σ3 nor in viral uncoating in itself. In addition, the μ1 protein simultaneously co-evolved, which apparently renders it more compatible with the modified σ1. Interestingly, the N198K substitution in σ1 appeared first during persistence establishment; this suggests that it is actually the critical determinant of adaptation to Vero cells, the other substitution (Q78P) appearing later on. The mutation in the σ1-encoding gene also results in a single amino acid substitution in a second protein (σ1s), a small protein found in a different but overlapping reading frame on the same gene [8]. As discussed below, this substitution is an important determinant of interferon sensitivity.
As substitutions at the level of σ1 appeared, other changes on μ1 also appeared. These substitutions (E89G and A114 V) are likely responsible for faster disassembly of the virion [9, 48]. However, again, these changes may not be desirable under natural conditions since they will result in an increased sensitivity to proteases in the gastrointestinal tract [9, 48]. To date, this is the only known example of a modification on μ1 in the context of viral persistence. Studies presently undergoing in the laboratory intend to better understand the significance, or not, of each of these substitutions [49]. Interestingly, one of these (E89G) has been independently examined by another group in the course of their studies of the loop formed by region 72-96 of the protein [50]. A substitution of amino acid 89 resulted in a small plaque phenotype in the presence of chymotrypsin, a phenotype that was also observed in our own studies [9, 48], despite the different genetic background of the virus used in these different studies. Similarly, while coevolution (or co-adaptation) of μ1 and σ1 was observed and is essential on VeroAV [9], subsequent works also indicated an effect of μ1 on the infectivity phenotype attributed to σ1. This underlines the importance of compatibility between these two proteins [51-53]. These observations are also in accordance with previous studies showing a role of μ1 on different phenotypes attributed to the S1 gene [54-58], although some of these may depend on the second reading frame encoding σ1s. Despite the lack of direct physical interaction between the two proteins, σ1 and μ1, an indirect effect on the structure of the viral particle could affect anchoring or exposure of σ1 at the surface of the viral particle [48].
Other cellular models
In addition to L929 cells, another murine fibroblast cell line has been studied for the ability of reovirus to establish persistent infection. These cells, the murine feral embryonic fibroblasts cell line, SC1, were initially chosen because they exhibit reduced host-cell protein synthesis inhibition and delayed killing upon infection [59]. When they are infected with a wild-type reovirus stock, a higher percentage of the cells can survive, compared to the situation observed with L929 cells. Consequently, persistence is more readily established but the phenomenon of host-cell coevolution can nevertheless be observed. Although, the exact cellular changes were not examined, cured cells could be obtained and they similarly exhibit resistance to wild-type virus and sensitivity to the PI virus recovered from these cells (unpublished data). Partial sequencing of the PI virus revealed an amino acid substitution at a different position on σ3 but close to those previously observed on PI viruses obtained from L929 cells [48] (figure 3); it turned out that the virus was also more resistant to the presence of uncoating inhibitors (unpublished data).
Various human cell types have also been examined such as lymphocytic cell lines Raji and CA46 as well as fibrosarcoma cells HT1080 [60-62]. An altered σ3, potentially resulting in a facilitated cleavage of the protein, is also selected in these cells (figure 3), but not always exclusively. In at least one case a truncated version of σ1 was also found (figure 4). This again underlines the importance of the two proteins σ3 and σ1, two critical proteins during virus entry in the host cell. Again, in all these cases, the complete sequence of the virus was not reported, thus complicating the interpretation.
A last case of viral persistence is in the CHO cell line (chinese hamster ovary cells) with selection of adapted PI virus and host-cell coevolution similar to the L929 cells situation [63]. These studies were not pursued for further characterization of the viruses and cells obtained. These results are also somewhat surprising, since later works have shown that these cells are poorly infected, due to the lack of protein receptor for the virus [64, 65]. Again, a difference in the exact nature of the cell lines used in these different studies likely explain these differences. It remains that the general principle of virus-cell coevolution also applies to this cell line.
The role of interferon in viral persistence
The role of the host-cell interferon response in reovirus persistence remains unclear. In SC1 cells, viral persistence was found to be associated with constant interferon secretion and activation of PKR [66, 67]. However, in these cells, interferon is not protective and this aspect has not been studied in other persistently infected cell lines. Viral persistence was also established in Vero cells that are deficient for interferon synthesis, the initial hypothesis was that this should allow virus evolution, independently of the constraint to maintain a certain level of resistance to interferon. As a result, it was expected that the resulting virus would be more sensitive to interferon and this was actually the case. The small nonstructural S1-encoded σ1s turned out to be a new determinant of viral sensitivity to interferon, independently of an effect on induction of interferon synthesis or release [8].
Other reflexions on the mechanisms of reovirus persistent infection
The establishment of viral persistence is facilitated when “attenuated” viruses or experimental conditions limiting viral replication, such as the addition of uncoating inhibitors, are used and the events related to viral entry are most important. Persistence establishment is also facilitated by using thermosensitive viruses or conditions where a constellation of mutant viruses are present from the beginning, for example by using high-passage virus stocks [31, 48, 68-71].
As for many eucaryotic viruses, the exact mechanism(s) leading to cell lysis remain(s) elusive and may involve more than one step of the viral multiplication cycle. Certain cell types are also known to be permissive for reovirus replication while remaining viable and releasing infectious virus. This was observed in the case of a bat cell line [22] as well as in endothelial and epithelial cells [2, 3]. The use of a vesicular transport mechanism cannot be excluded as a mechanism for this release without lysis, especially considering recent observations showing remodelling of internal cellular membranes at the level of viral factories in cells infected by reovirus [72]. Another possibility is that reovirus can exploit the autophagic machinery. In the last few years, examples have been accumulating concerning the use of autophagy by viruses, cytolytic or not, for their assembly or release from infected cells [73-77]. More recently, the participation of autophagy in the reovirus oncolytic activity, at least in some cell types, has also been reported [78, 79]. It is thus possible that such a transport by the autophagic machinery could be exploited by reovirus for a non-cytolytic release, as happens from persistently infected cells. In fact, electron microscopic examination of persistently infected cells has shown the presence of viral particles in vesicles, some of them morphologically reminiscent of autophagosomes [17, 23, 29, 66]. However, the exact nature of these vesicles has not been clearly established. A permanent activation of PKR in reovirus persistence has also been previously noticed [66]; such a PKR activity could lead to increased autophagy [80], thus contributing to viral release independently of cell lysis.
In all described examples, reovirus persistent infection requires a constant viral reinfection. This implies that the virus achieves a complete replication cycle and that released virions are infectious. It explains that upon an exposure for a relatively long time to neutralizing antibodies, cells are cured to an undetectable level of viral production, while the removal of the antibody does not lead to a rebound of viral production.
Earlier works suggested a model of viral persistence in which establishment and maintenance are two distinct phenomena relying on different and stepwise genetic alterations on PI viruses. In the first model, changes on σ3 were considered as essential to the establishment of persistence, as discussed in the above sections. In contrast, maintenance seemed to rely more on changes at the level of the σ1 protein [32, 71]. It should be mentioned that some of these studies suggested that λ2 could be involved in the establishment of viral persistence by increasing the frequency of mutations in the viral population used for the infection leading to persistence [71]. Although the abundant presence of mutations in the viral population could certainly contribute to the establishment of persistence, a role for the λ2 protein seems unlikely; the idea that this protein could act as the viral polymerase is not compatible with our current knowledge on the transcription and replication of the viral genome [11].
In the Vero cells model the amino acid substitution on σ1 allowing adaptation of the virus to the cells, first appears during the establishment of persistence. Later, the change on σ1s responsible for interferon sensitivity and the changes to μ1 will appear [48]. It is thus likely that the changes described as responsible for “maintenance” are less directly involved in persistence as such but are rather an indirect consequence of viral growth for an extended period of time in a given cell type. The order and the nature of the changes observed in L929 and Vero cells are summarized schematically in figure 5.
In vivo importance
Few studies have addressed reovirus persistence in vivo. In the past, the presence of mutant viruses selected in vivo was shown. It also appeared that hybridoma cells obtained following viral inoculation are sometimes persistently infected and can be cured in vitro by neutralizing antiserum treatment [81, 82]. These different PI viruses were not further studied.
From another point of view, inoculation of PI viruses recovered from L929 cells seemed to indicate an increased virulence of these viruses, at least by intracerebral inoculation [83, 84]. Finally, tumour cell lines cured from persistent infection in vitro remained sensitive to the wild-type virus in the context of tumours in vivo, even though they are in fact resistant in vitro, whereas persistently-infected cells are unable to form tumours under these conditions [60, 61]. Clearly, these different aspects of in vivo persistence deserve further studies.
Viral persistence as a tool to select new reoviruses
The in vitro evolution or adaptation of viruses to a given cell type is not only very informative from a fundamental point of view but represents a potential tool to produce new viruses. The selection or construction of new reoviruses better adapted as virotherapeutic agents, gene vectors, vaccines, or oncolytic viruses, has been proposed by many authors [6, 7, 26, 62, 58-89]. In the context of viral persistence, one example of a PI virus selected on a tumour cell line was shown to be attenuated on normal cells without altering its infectivity or cytolytic potential on transformed cells [62]. In this particular case, a deletion of a part of the σ1 protein was observed rather than amino acids substitutions.
Serial passage or the selection of viral clones are two possible approaches to select new viruses harbouring useful phenotypes [26, 89, 90]. However, the long-term interaction needed to establish and maintain persistence represents an impressive number of viral cycles that cannot be readily achieved otherwise by these other approaches. Furthermore, the development of cellular resistance is likely a driver of viral evolution in this context, compared to serial passage, although this last approach probably results in a different type of selective pressure.
Obtaining PI viruses from different cell types, coupled with genome sequencing of these viruses and confirmation of the importance of various substitutions using reverse genetics, could lead to a better understanding of the persistence phenomena and virus-cell coevolution. It will certainly be of interest to obtain viruses at different times during the establishment of persistence to better determine the nature of the changes that are directly related and the phenomenon of coevolution in various cellular contexts.
Conclusions
Despite the fact that reovirus persistence was described more than 40 years ago, the study of underlying mechanisms remains pertinent. First of all, such studies could allow a better fundamental understanding of virus biology in various cell types. Secondly, these studies could lead, on a longer term, to the identification of new viral variants that could be used in anticancer virotherapy or in other applications.
Acknowledgments
The work carried out in the author's laboratory is presently supported by the Natural Sciences and Engineering Research Council of Canada (NSERC). The author also wishes to thank all staff, collaborators, as well as numerous students and trainees, for their contributions to the work performed in the laboratory over the years.
Conflict of interests
The author has no conflict of interest concerning this article.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License