Néphrologie & Thérapeutique
MENUAdult IgA vasculitis (Henoch-Schönlein purpura) Volume 15, supplement 1, Avril 2019

- Key words: IgA vasculitis, Small vessel vasculitis, Purpura, IgA glomerulonephritis, Adults
- DOI : 10.1016/j.nephro.2019.02.001
- Page(s) : 13-20
- Published in: 2019
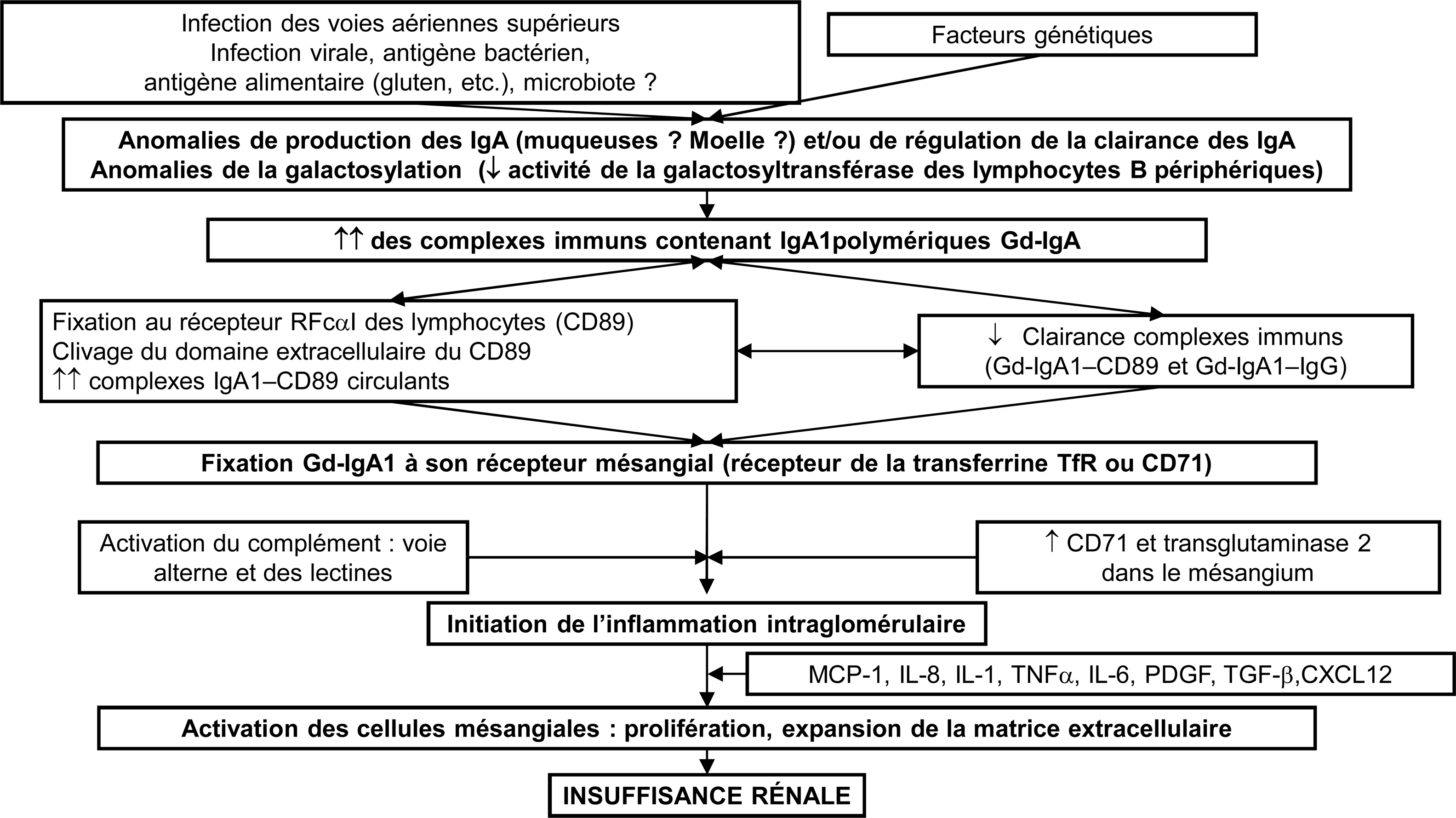
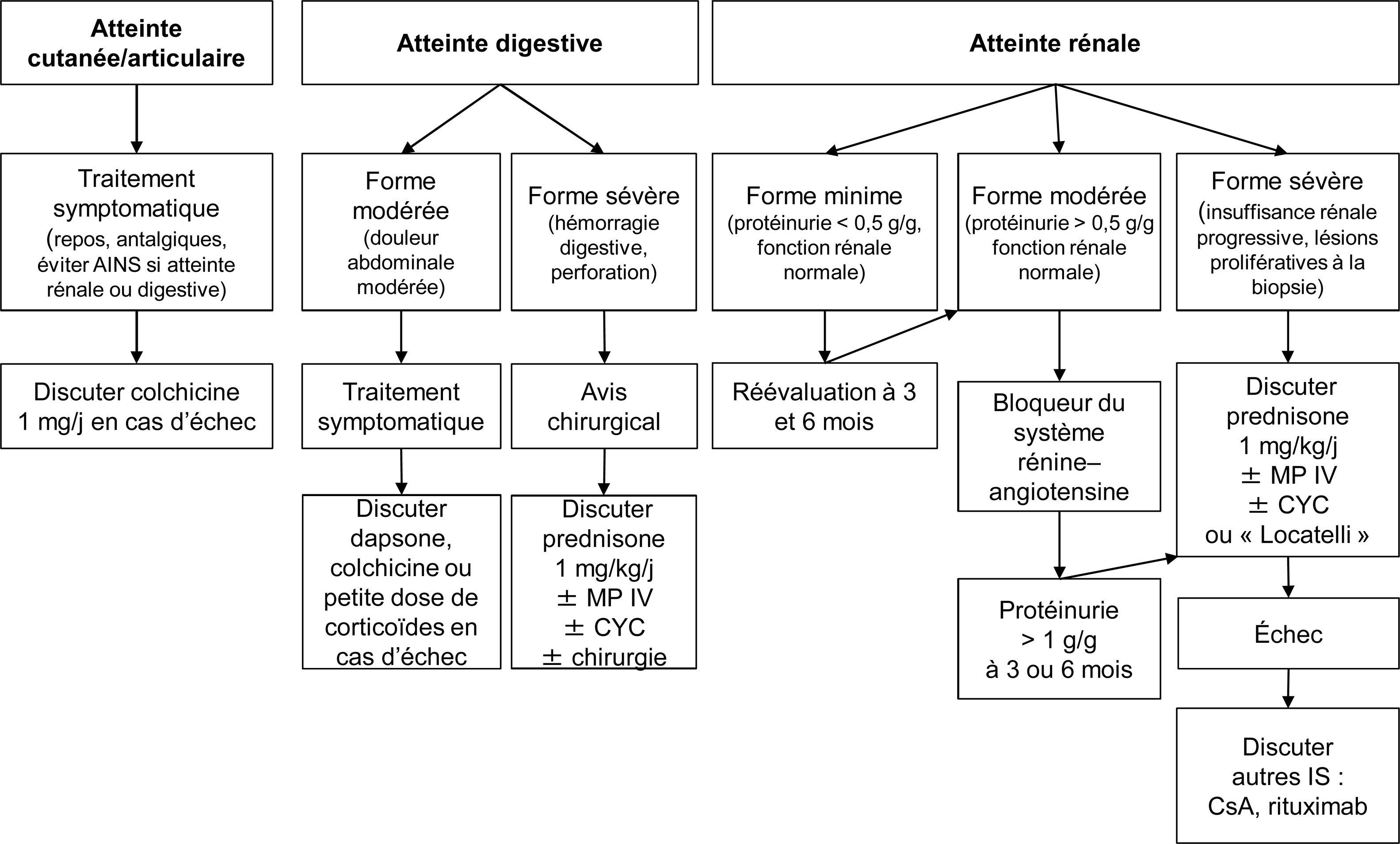
La vascularite à IgA, anciennement purpura rhumatoïde est une vascularite systémique des petits vaisseaux à dépôts d’immunoglobulines A (IgA). Elle est beaucoup plus fréquente chez l’enfant que chez l’adulte (150 à 200 pour 1). La prévalence de la vascularite à IgA chez l’adulte n’est pas connue et son incidence est estimée à 1 par million. La maladie de l’adulte semble en effet différer de celle de l’enfant par l’incidence et la gravité des manifestations cliniques. La vascularite à IgA est caractérisée par l’association d’un purpura vasculaire cutané à des signes articulaires et gastro-intestinaux. Une atteinte rénale s’associe parfois à ces signes. Il s’agit alors d’une glomérulonéphrite à dépôts mésangiaux d’IgA. Plus rarement des localisations neurologiques, pulmonaires, cardiaques ou urogénitales peuvent s’observer. Sa physiopathologie demeure inconnue mais les IgA joueraient un rôle central. Lorsque la symptomatologie est peu sévère seul un traitement symptomatique est conseillé. À l’opposé, dans les formes digestives ou rénales sévères, des traitements plus agressifs, associant le plus souvent, des corticostéroïdes à un immunosuppresseur ont été proposés. Leur efficacité est en cours d’évaluation. Le pronostic à court terme de la maladie dépend de la sévérité de l’atteinte digestive et à long terme, de la sévérité de l’atteinte rénale. Les études ayant un suivi suffisamment prolongé montrent qu’un tiers des malades adultes évoluent vers l’insuffisance rénale terminale, comme pour la néphropathie à IgA. De nombreux auteurs suggèrent d’ailleurs que la néphropathie à IgA et la vascularite à IgA seraient deux entités de la même maladie.
IgA vasculitis is a systemic vasculitis affecting small vessels. IgA vasculitis usually affect children whereas it is rare in adults (150 to 200 for 1) in which the disease is often more serious with more frequent and severe nephritis. Prevalence of adult IgA vasculitis is unknown and its annual incidence is 1 in 1 million. The dominant clinical features include cutaneous purpura, arthritis and gastrointestinal symptoms. Sometimes nephritis can add, typically as glomerulonephritis with IgA mesangial deposits. Pulmonary, cardiac, genital and neurological symptoms have also been observed. Although the cause is unknown, it is clear that IgA plays a pivotal role in the immunopathogenesis of IgA vasculitis. Only symptomatic treatment is advised in case of self-limited disease. Treatment of severe IgA vasculitis, nephritis or gastrointestinal manifestations, is not established but some studies, which need to be confirmed, reported the benefit of corticosteroids combined with immunosuppressive drugs. Short-term outcome depends of the severity of the gastro-intestinal manifestations. The long-term prognosis is heavily dependent on the presence and severity of nephritis. Studies with prolonged follow-up show up to one third of adult patients reaching end stage renal failure, as for IgA nephropathy. Some authors even suggest that IgA nephropathy and IgA vasculitis would be the same disease.