Néphrologie & Thérapeutique
MENUTreatment of AL amyloidosis Volume 15, supplement 1, Avril 2019

- Key words: AL amyloidosis, Treatment, Free light chains, Bortezomib, Melphalan, Dexamethasone, Cyclophosphamide, Haematologic response
- DOI : 10.1016/j.nephro.2019.03.002
- Page(s) : 115-21
- Published in: 2019
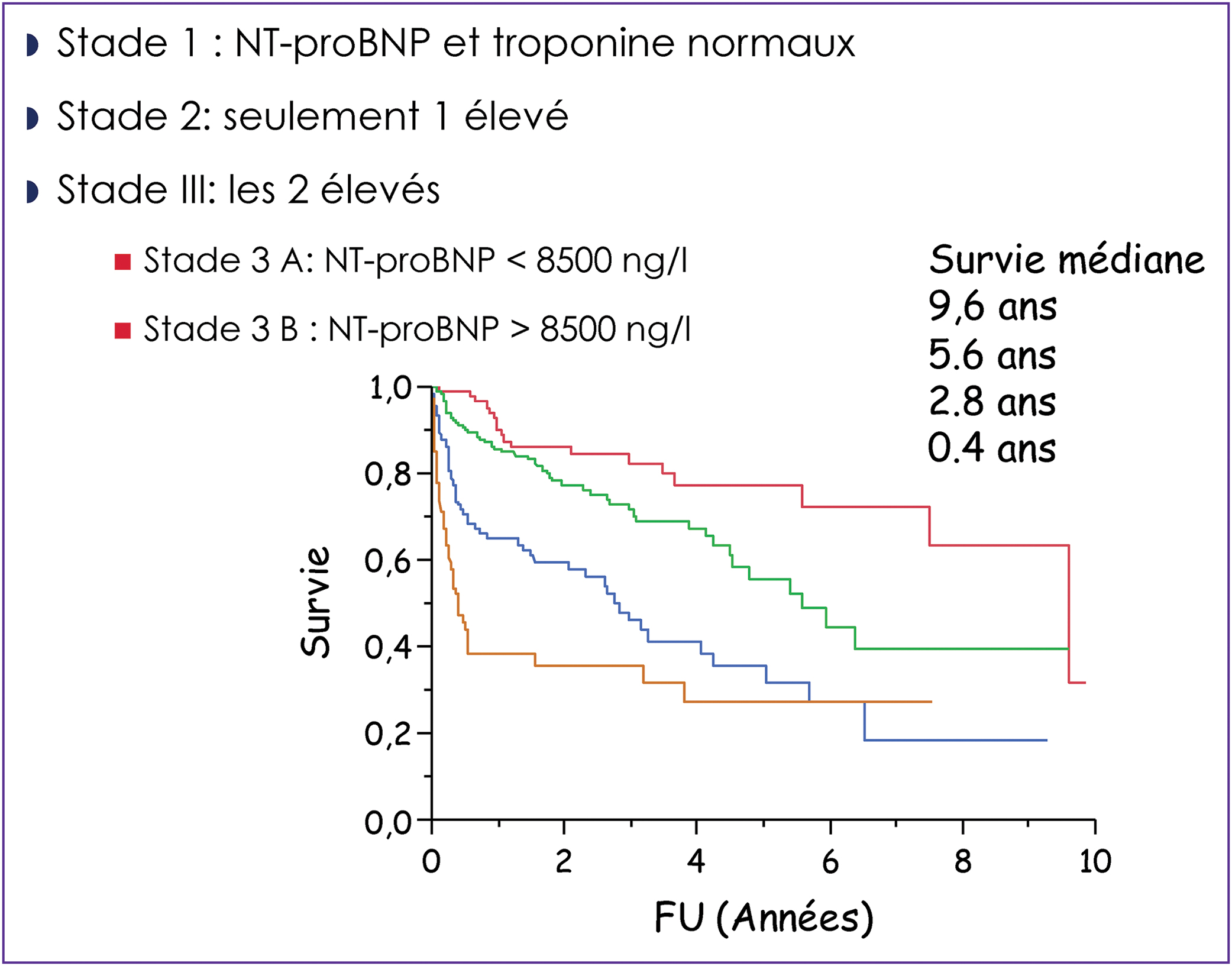
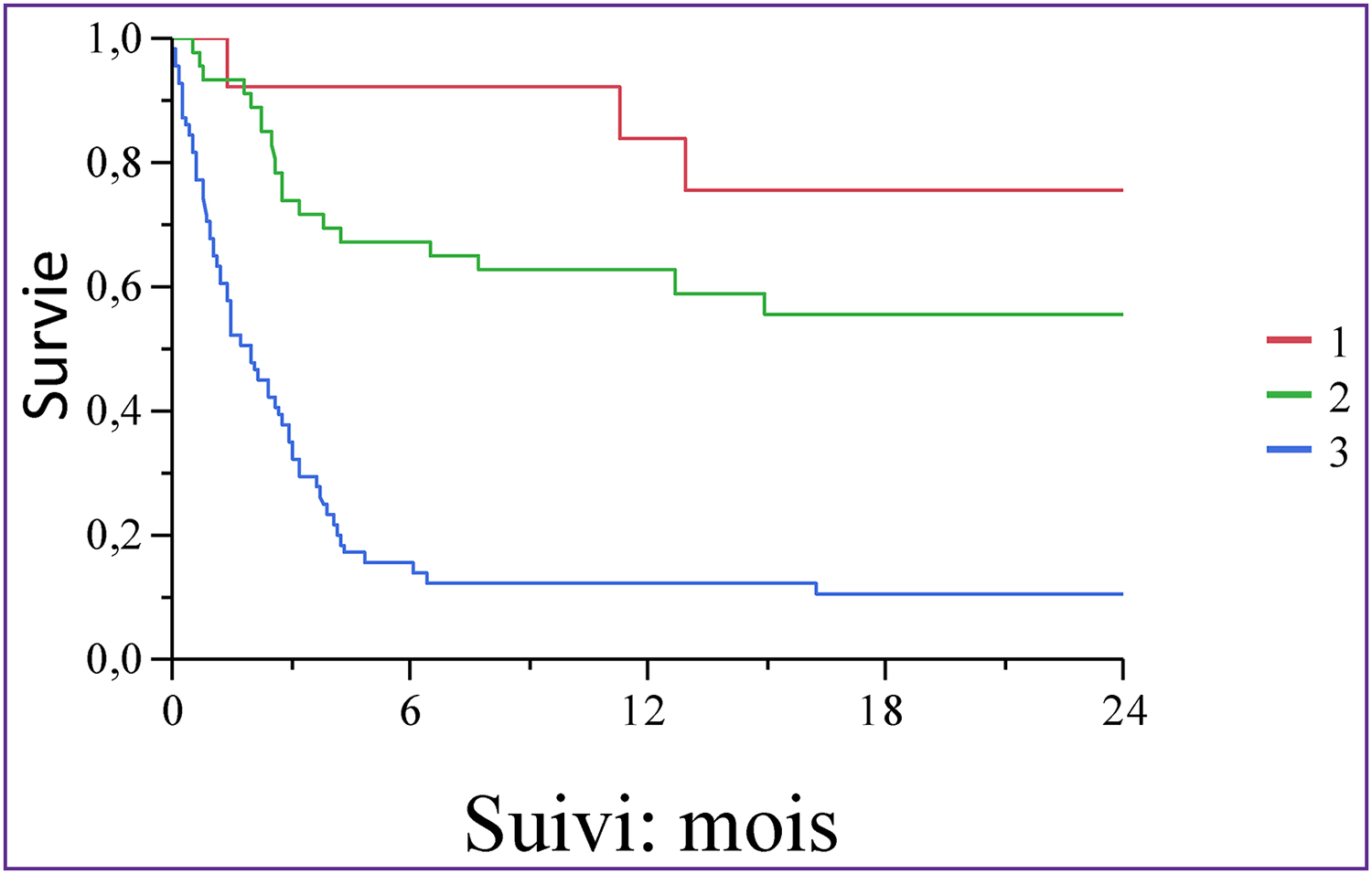
L’amylose AL est liée au dépôt tissulaire de fibrilles composées de chaînes légères monoclonales d’immunoglobulines. Son diagnostic est histologique montrant sur des biopsies le plus souvent non invasives des dépôts extracellulaires amorphes colorés par le rouge Congo, constitués de chaînes légères, le plus souvent lambda. La présentation clinique est extrêmement polymorphe en raison du grand nombre d’organes pouvant être atteints. Les manifestations les plus fréquentes, concernant chacune environ deux tiers des patients, sont l’atteinte rénale, caractérisée le plus souvent par un syndrome néphrotique, et la cardiopathie restrictive. Le traitement repose sur une chimiothérapie visant à faire disparaître le clone médullaire producteur de la chaîne légère monoclonale. Il est guidé par la gravité de la maladie, établie à l’aide des marqueurs cardiaques. Son efficacité doit être régulièrement évaluée par le dosage sérique des chaînes légères libres monoclonales. Les traitements de référence actuels, associant un alkylant, de fortes doses de dexaméthasone, et le plus souvent un inhibiteur du protéasome, sont efficaces chez la majorité des patients. Le pronostic dépend de l’importance des atteintes initiales, particulièrement cardiaque, et est très fortement influencé par la réponse hématologique au traitement. Celui-ci doit être modifié chez les patients non répondeurs, d’autant plus rapidement que la maladie, en particulier cardiaque, est sévère. L’introduction d’un immunomodulateur ou d’un anticorps ciblant les plasmocytes doit alors être considérée. Le traitement des patients avec une atteinte cardiaque grave reste difficile. Les stratégies visant à accélérer l’élimination des dépôts, malgré des résultats préliminaires décevants, devraient permettre d’améliorer encore le pronostic de cette maladie.
AL amyloidosis is caused by the conversion of monoclonal immunoglobulin light chains into amyloid fibrillar aggregates that deposit in tissue and lead to organ dysfunction. Diagnosis is histological and relies primarily on non-invasive biopsies, showing Congo red-positive amorphous deposits containing immunoglobulin light chains, most commonly of lambda isotype. The clinical presentation is extremely polymorphous, due to the large number of organs that can be affected by the disease. The kidneys and the heart are most frequently involved organs, in about two thirds of patients each, responsible for nephrotic syndrome and restrictive cardiomyopathy. Treatment is based on chemotherapy aimed at eliminating the medullary clone producing the pathogenic monoclonal light chains. It is guided by risk assessment, based on the serum levels of cardiac biomarkers. Its effectiveness must be regularly assessed by the serum free light chain assay. Current reference regimens combine an alkylating agent, with high doses of dexamethasone and most often a proteasome inhibitor. They are effective in the majority of patients. The overall prognosis depends on the importance of the initial severity of organ involvement, particularly the heart, and is strongly influenced by the haematological response. The treatment must be rapidly modified in non-responders, especially in those with severe cardiac disease, with the introduction of immunomodulatory drugs and antibodies targeting plasma cells. However, effective therapies for patients with the more severe amyloid cardiopathy are an unmet need. Strategies directly accelerating the removal of amyloid deposits, despite disappointing preliminary results, could further improve the prognosis of this disease.