Néphrologie & Thérapeutique
MENUHemolytic uremic syndrome in adults Volume 6, issue 4, Juillet 2010

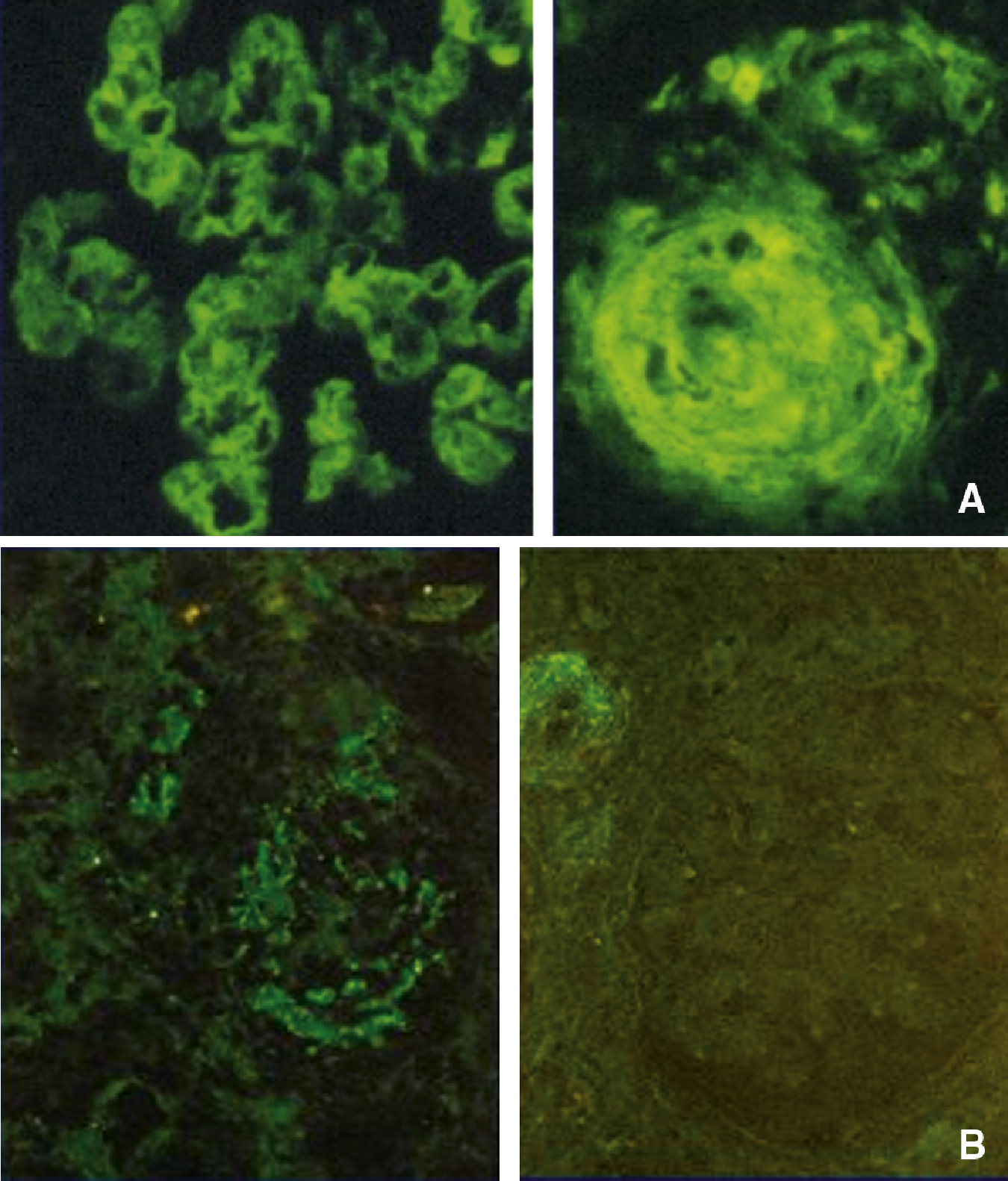
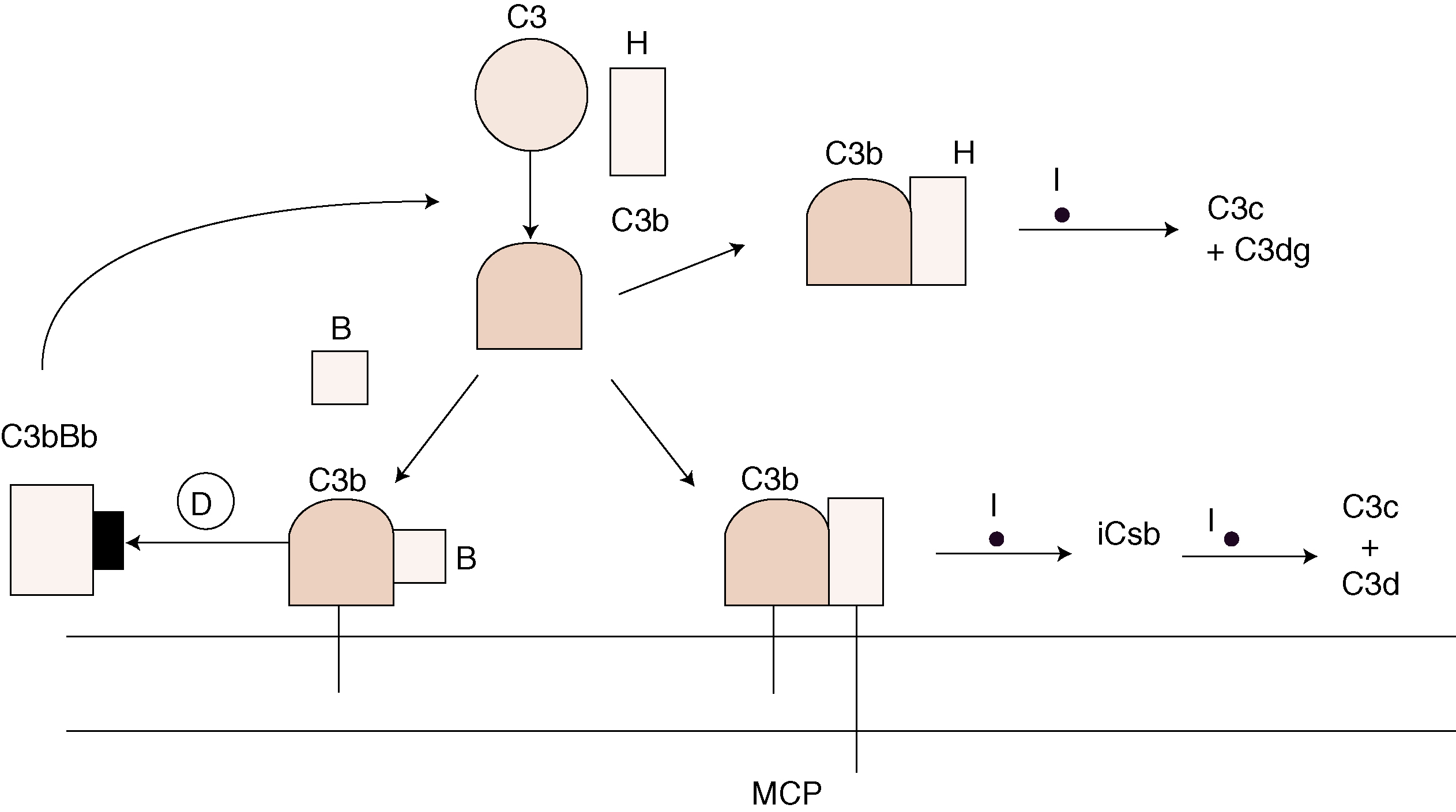
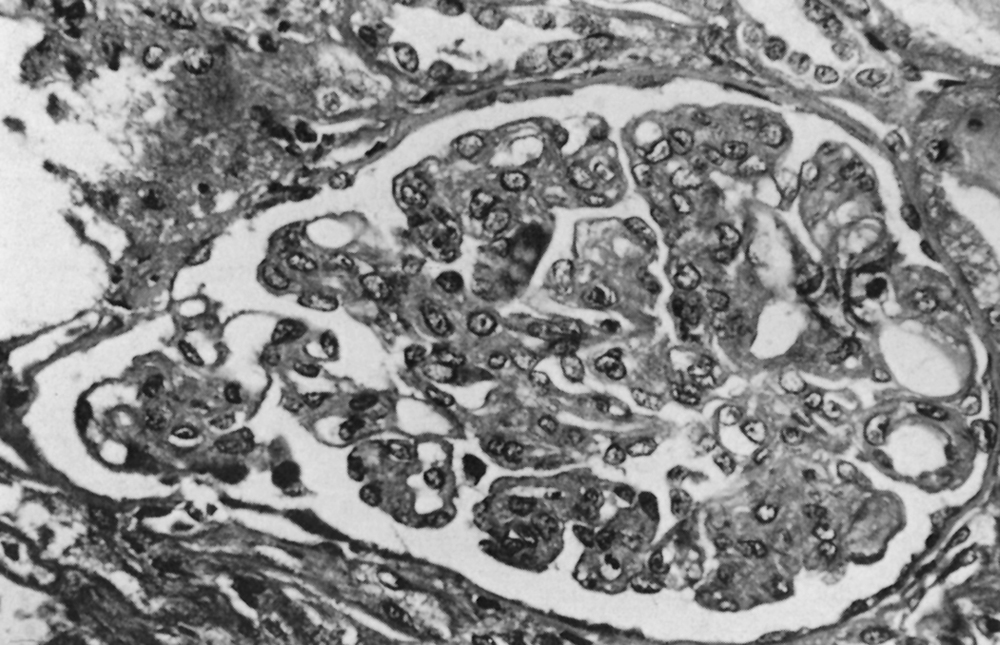
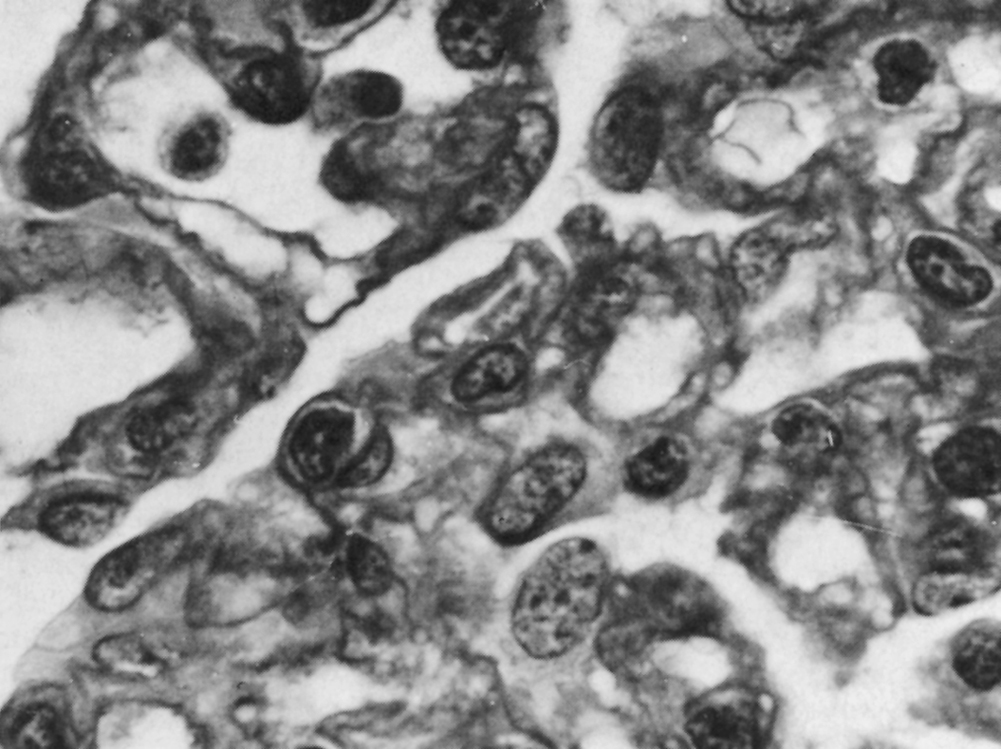
- Key words: Hemolytic uremic syndrome, Shigatoxin, Renal failure, Factor H, Plasma exchanges
- DOI : 10.1016/j.nephro.2010.03.002
- Page(s) : 258-71
- Published in: 2010
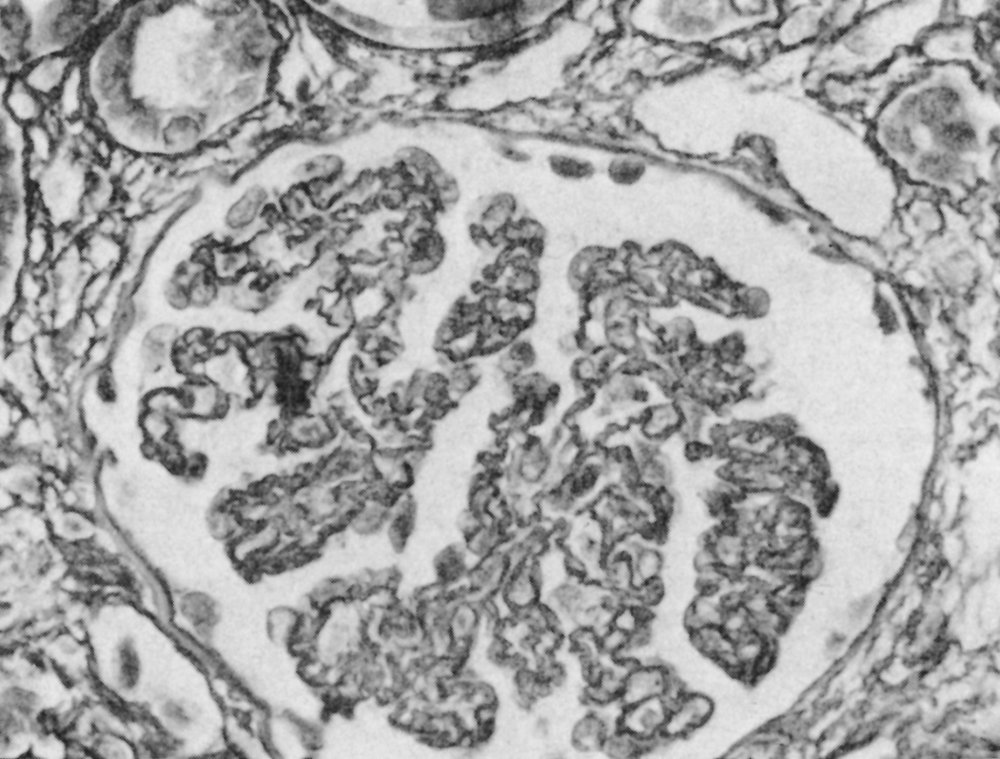
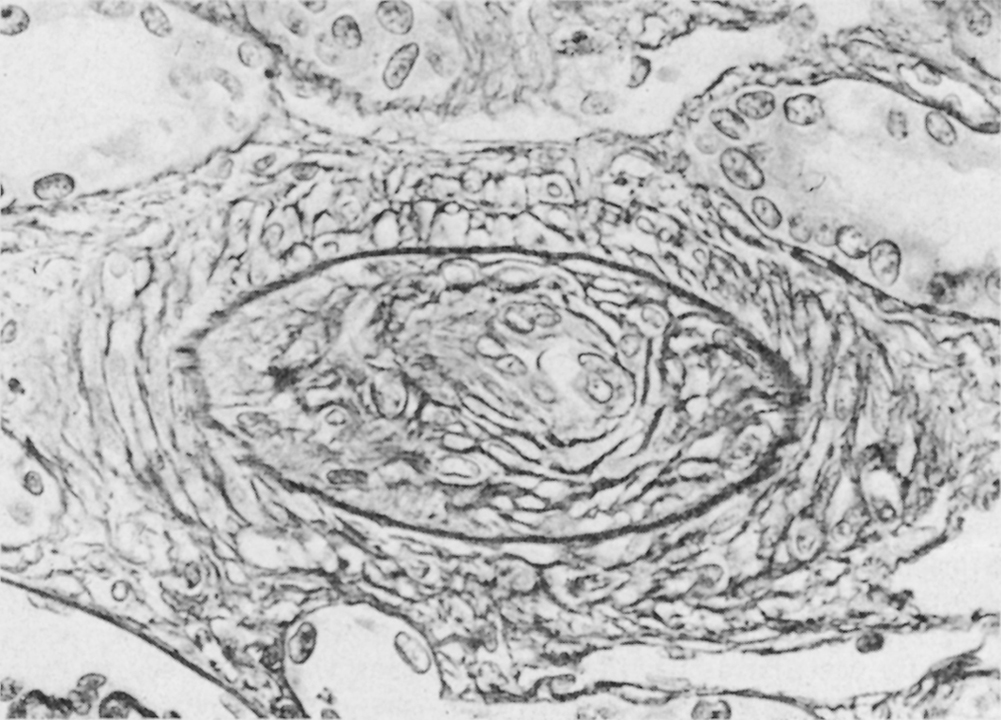
Le syndrome hémolytique et urémique (SHU) est dû à une microangiopathie thrombotique intrarénale, entraînant une hypertension artérielle et une insuffisance rénale aiguë (IRA). Sa pathogénie comprend une lésion/activation de l’endothélium, essentiellement au niveau rénal, induite par des entérotoxines bactériennes, des médicaments toxiques, ou des autoanticorps. Une hyperactivation de la voie alterne du complément en rapport avec un déficit hétérozygote en protéines régulatrices (facteur H, facteur I ou protéine MCP) peut aussi être associée à un SHU. Plus rarement, les microthromboses sont liées à un déficit acquis ou congénital en protéase du facteur de von Willebrand. L’anémie hémolytique mécanique avec présence de schizocytes, la thrombopénie sans coagulation intravasculaire disséminée, et l’insuffisance rénale sont caractéristiques. Dans les SHU typiques, une diarrhée prodromique, volontiers sanglante, est observée, en rapport avec une infection par une entérobactérie pathogène, souvent le E. coli O157 : H7. Le SHU peut aussi être observé dans le post-partum, et est alors en rapport avec un déficit en facteur H. Il peut aussi compliquer un traitement par mitomycine C, gemcitabine, ciclosporine A, ou tacrolimus, et plus récemment le bevacizumab, un anticorps anti-VEGF. Les SHU atypiques, sans diarrhée prodromique, qu’ils soient sporadiques ou familiaux, peuvent révéler un déficit hétérozygote en facteur H, facteur I ou protéine MCP. Des formes autoimmunes avec anticorps anti-facteur H ont aussi été décrites, ainsi que des mutations avec gain de fonction du facteur B ou du C3. Plus récemment des mutations de la thrombomoduline, qui participe aussi à la régulation de la voie alterne, ont été décrites dans des formes familiales de SHU. Certains SHU compliquent l’évolution de néphropathies chroniques : néphroangiosclérose, glomérulonéphrites chroniques, néphrite radique, sclérodermie, lupus érythémateux disséminé, syndrome des antiphospholipides. Le pronostic s’est amélioré, avec une survie des malades de plus de 85 % à 1 an. L’insuffisance rénale chronique séquellaire est observée dans 20 à 65 % des cas. Les perfusions de plasma frais ou les échanges plasmatiques permettent le plus souvent de corriger l’hémolyse et la thrombopénie. La corticothérapie est discutée, ainsi que les immunosuppresseurs dans les formes autoimmunes réfractaires ou à rechute, liées parfois à un déficit en protéase du facteur de von Willebrand. Un anticorps anti-C5, l’eculizumab, est actuellement à l’essai mais semble être très efficace dans les formes liées à une hyperactivation de la voie alterne du complément. En cas d’insuffisance rénale terminale, la transplantation rénale est possible mais le risque de récidive, quasi nul dans les formes postinfectieuses, est de 70 à 80 % dans les formes liées à un déficit en facteur H, I ou en protéine MCP.
Hemolytic uremic syndrome (HUS) is related to a renal thrombotic microangiopathy, inducing hypertension and acute renal failure (ARF). Its pathogenesis involves an activation/lesion of microvascular endothelial cells, mainly in the renal vasculature, secondary to bacterial toxins, drugs, or autoantibodies. An overactivation of the complement alternate pathway secondary to a heterozygote deficiency of regulatory proteins (factor H, factor I or MCP) or to an activating mutation of factor B or C3 can also result in HUS. Less frequently, renal microthrombi are due to an acquired or a constitutional deficiency in ADAMTS-13, the protease cleaving von Wilebrand factor. Hemolytic anemia with schistocytes, thrombocytopenia without evidence of disseminated intravascular coagulation, and renal failure are consistently found. In typical HUS, a prodromal diarrhea, with blood in the stools, is observed, related to pathogenic enterobacteria, most frequently E. Coli O157:H7. HUS may also occur in the post partum period, and is then related to a factor H or factor I deficiency. HUS may also occur after various treatments such as mitomycin C, gemcitabine, ciclosporin A, or tacrolimus, and as reported more recently bevacizumab, an anti VEGF antibody. Atypical HUS are not associated with diarrhea, may be sporadic or familial, and can be related to an overactivation of the complement alternate pathway. More recently, some of them have been related to a mutation of thrombomodulin, which also regulates the alternate pathway of complement. In adults, several HUS are encountered in the course of chronic nephropathies: nephroangiosclerosis, chronic glomerulonephritis, post irradiation nephropathy, scleroderma, disseminated lupus erythematosus, antiphospholipid syndrome. Overall the prognosis of HUS has improved, with a patient survival greater than 85% at 1year. Chronic renal failure is observed as a sequella in 20 to 65% of the cases. Plasma infusions and plasma exchanges are effective in most of the cases to treat hemolysis and thrombocytopenia. Steroid therapy is debated, as well as immunosuppressive drugs, including rituximab, in autoimmune forms. A new monoclonal anti-C5 antibody is tested, and seems to be effective in atypical HUS with abnormal complement alternate pathway activation. If terminal renal failure occurs, renal transplantation can be performed but the risk of recurrence, which very low in post infectious forms of HUS, is about 70 to 80% in genetic forms of complement regulatory protein deficiency.