Néphrologie & Thérapeutique
MENUBartter–Gitelman syndromes Volume 16, issue 4, Juillet 2020

- Key words: Renal tubular disease, Hypokaliemia, Hypomagnesemia, Polyhydramnios, Gitelman syndrome, Impaired salt reabsorption
- DOI : 10.1016/j.nephro.2020.06.001
- Page(s) : 233-43
- Published in: 2020
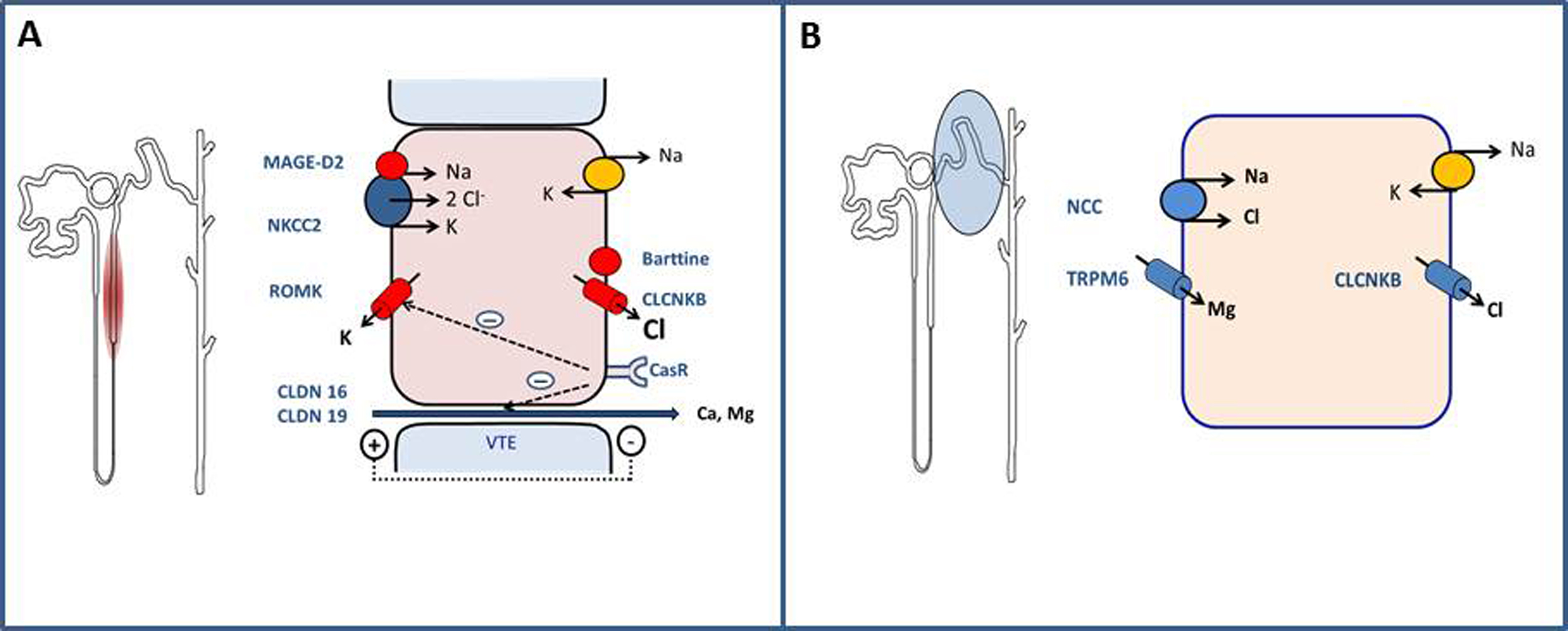
Les syndromes de Bartter–Gitelman sont des tubulopathies héréditaires avec perte en sel, caractérisées par une hypokaliémie avec alcalose métabolique hypochlorémique due à la stimulation du système rénine-angiotensine-aldostérone. Le syndrome de Bartter mime la prise de furosémide, associant un syndrome de perte en sel et une hypercalciurie avec défaut de concentration des urines. Les premiers symptômes peuvent être anténataux avec un -hydramnios suivi d’une déshydratation néonatale sévère, ou plus tardifs dans l’enfance pour le syndrome de Bartter classique. Le syndrome de Gitelman mime la prise de thiazidique associant au syndrome de perte en sel, une hypomagnésémie et une hypocalciurie (rapport calcium/créatinine inférieur à 0,20mmol/mmol), sans défaut de concentration des urines. Sa présentation n’est jamais anté- ou néonatale. En plus d’épisodes de déshydratation aiguë, le tableau clinique des syndromes de Bartter–Gitelman inclut des crises de tétanie, un retard de croissance, une fatigue chronique souvent intense, des crampes musculaires, des myalgies et des vertiges parfois rotatoires (données personnelles non publiées). La complication majeure du syndrome de Gitelman en termes de pronostic fonctionnel est la chondrocalcinose, tandis que seul le Bartter se complique parfois d’une insuffisance rénale. Le traitement des nouveau-nés atteints du syndrome de Bartter doit se concentrer sur la correction des troubles hydro-électrolytiques dans une unité de soins intensifs néonatale avec introduction secondaire d’indométacine. Ces syndromes nécessitent une supplémentation orale à vie en potassium, sodium (Bartter/Gitelman) et magnésium (Gitelman). Les antagonistes de l’aldostérone et diurétiques épargneurs de potassium peuvent aider à corriger l’hypokaliémie, mais aggravent la déplétion sodée.
Bartter–Gitelman syndromes are rare inherited autosomal recessive salt-losing tubulopathies characterized by severe and chronic hypokalemia associated with metabolic alkalosis and secondary hyperaldosteronism. Bartter syndrome results from a furosemide-like defect in sodium reabsorption in the Henle's loop leading to hypercalciuria and defect in urinary concentration capacity. The antenatal Bartter syndrome is defined by polyhydramnios and an infantile polyuria with severe dehydration whereas classic Bartter syndrome appears during childhood or adulthood. Gitelman syndrome is a thiazide-like salt-losing tubulopathy. It is associated with hypomagnesemia, hypocalciuria without defect in urinary concentration capacity. The diagnosis is most often made in adolescents or adults. Clinical symptoms include tetany, delay in the height-weight growth curves, chronic tiredness, muscle weakness, myalgia and vertigo. Nephrocalcinosis in Bartter syndrome could lead to chronic kidney disease. Antenatal Bartter syndrome requires hospitalization in intensive care unit to manage the severe newborn dehydration. Chondrocalcinosis is the major complication in the Gitelman syndrome. The corner stones of treatment is the fluid and electrolyte management Bartter and Gitelman syndromes need lifelong oral supplementations of potassium, salt (Bartter) and magnesium (Gitelman). Indomethacin is efficient to reduce water and electrolyte loss in Bartter. In Gitelman, potassium-sparing diuretics may be helping for severe hypokaliemia but they will reinforce hypovolemia.