Néphrologie & Thérapeutique
MENUAlport syndrome: Hereditary nephropathy associated with mutations in genes coding for type IV collagen chains Volume 12, issue 7, Décembre 2016

- Key words: COL4A3, COL4A4, COL4A5, COL4A6, Collagen IV, Diffuse oesophageal leiomyomatosis, Glomerular basement membrane, Alport syndrome, Macrothrombocytopathy
- DOI : 10.1016/j.nephro.2016.09.001
- Page(s) : 544-51
- Published in: 2016
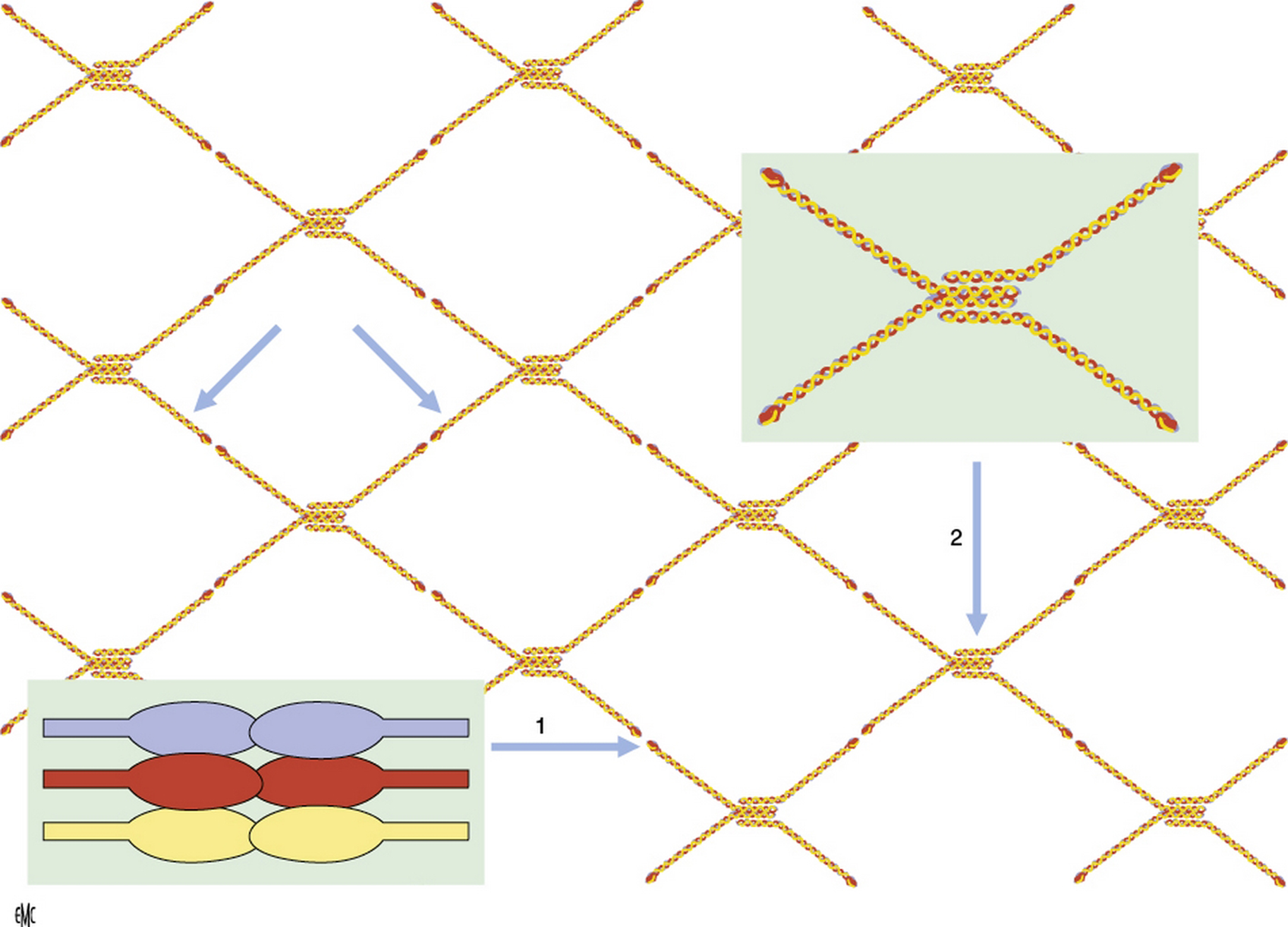
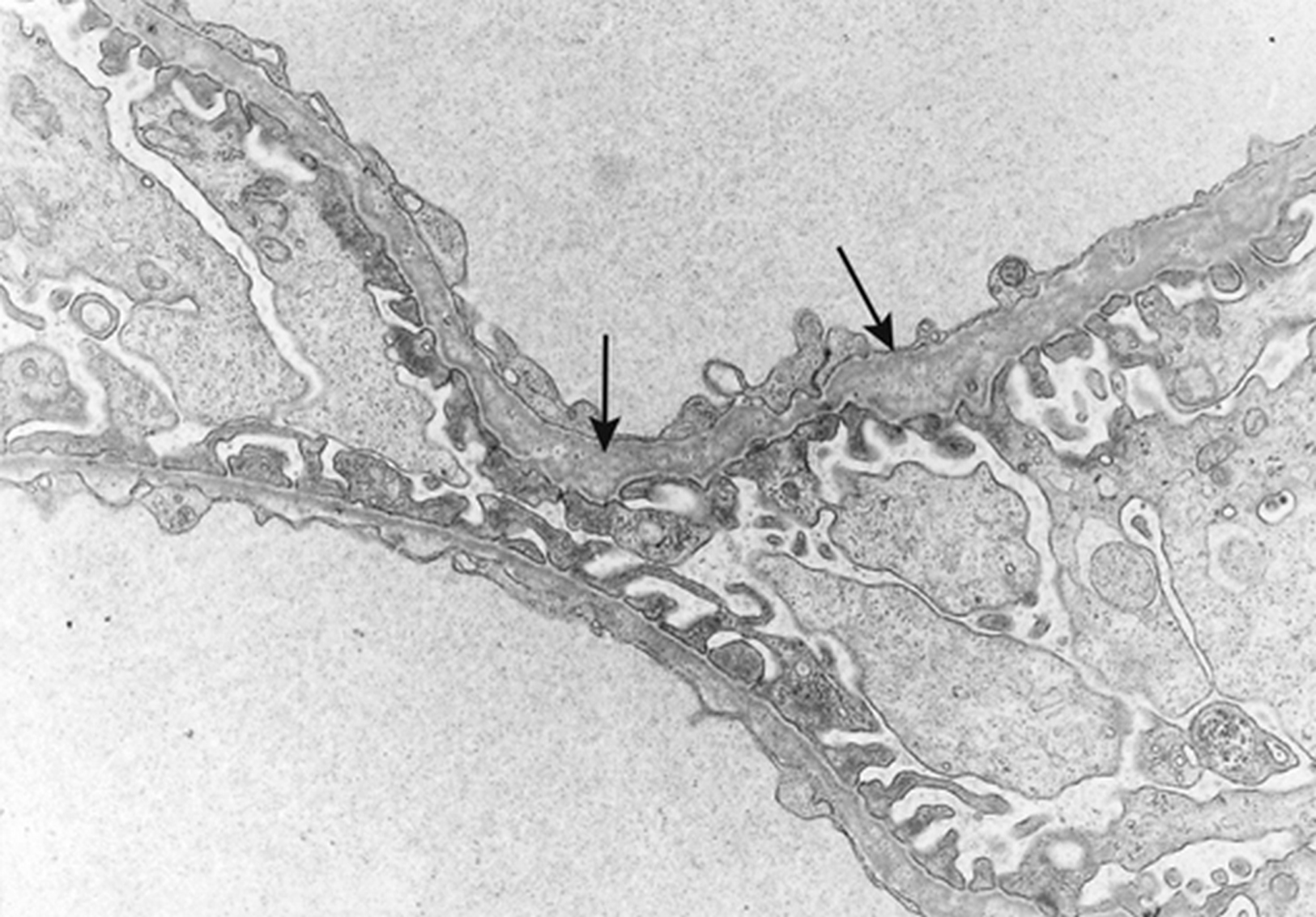
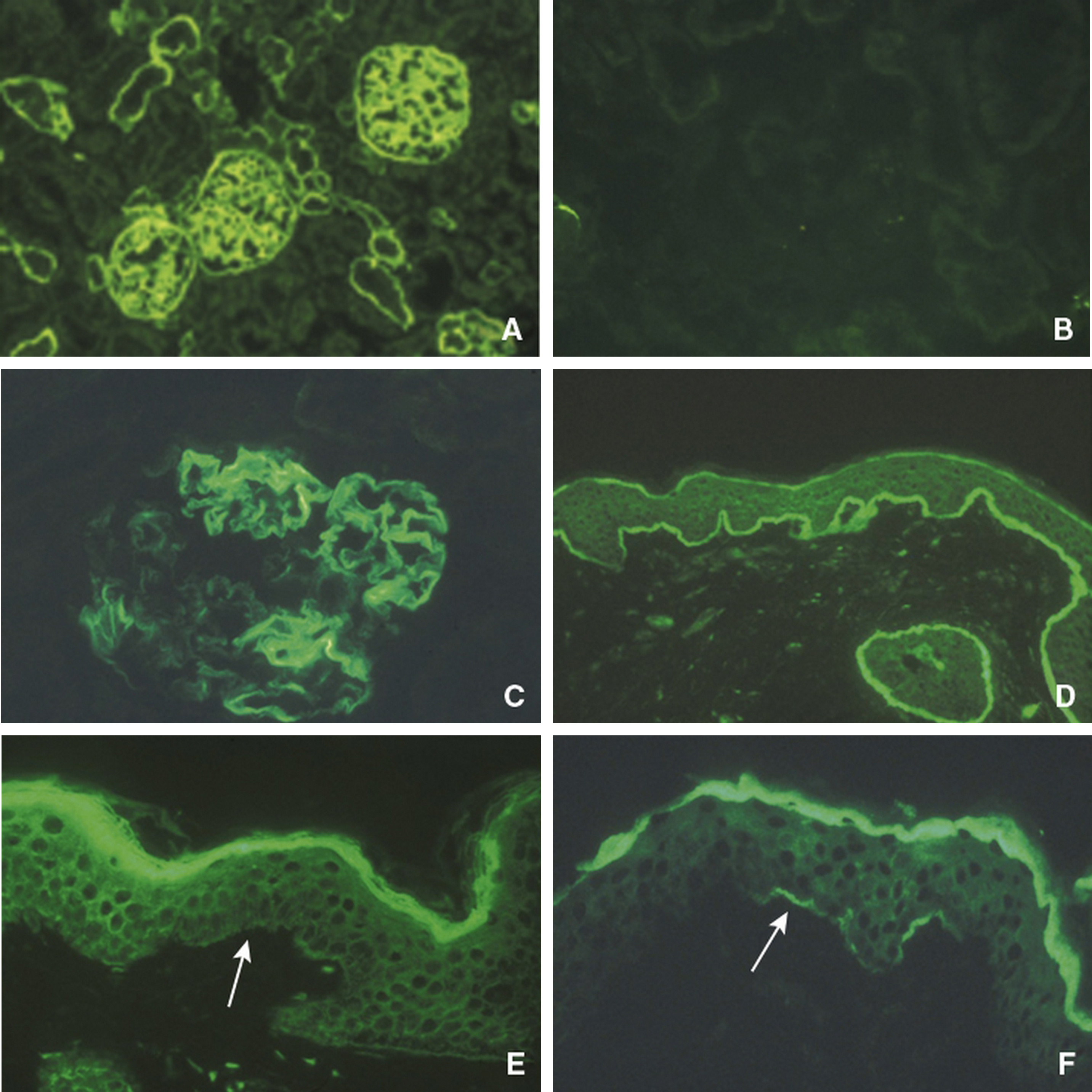
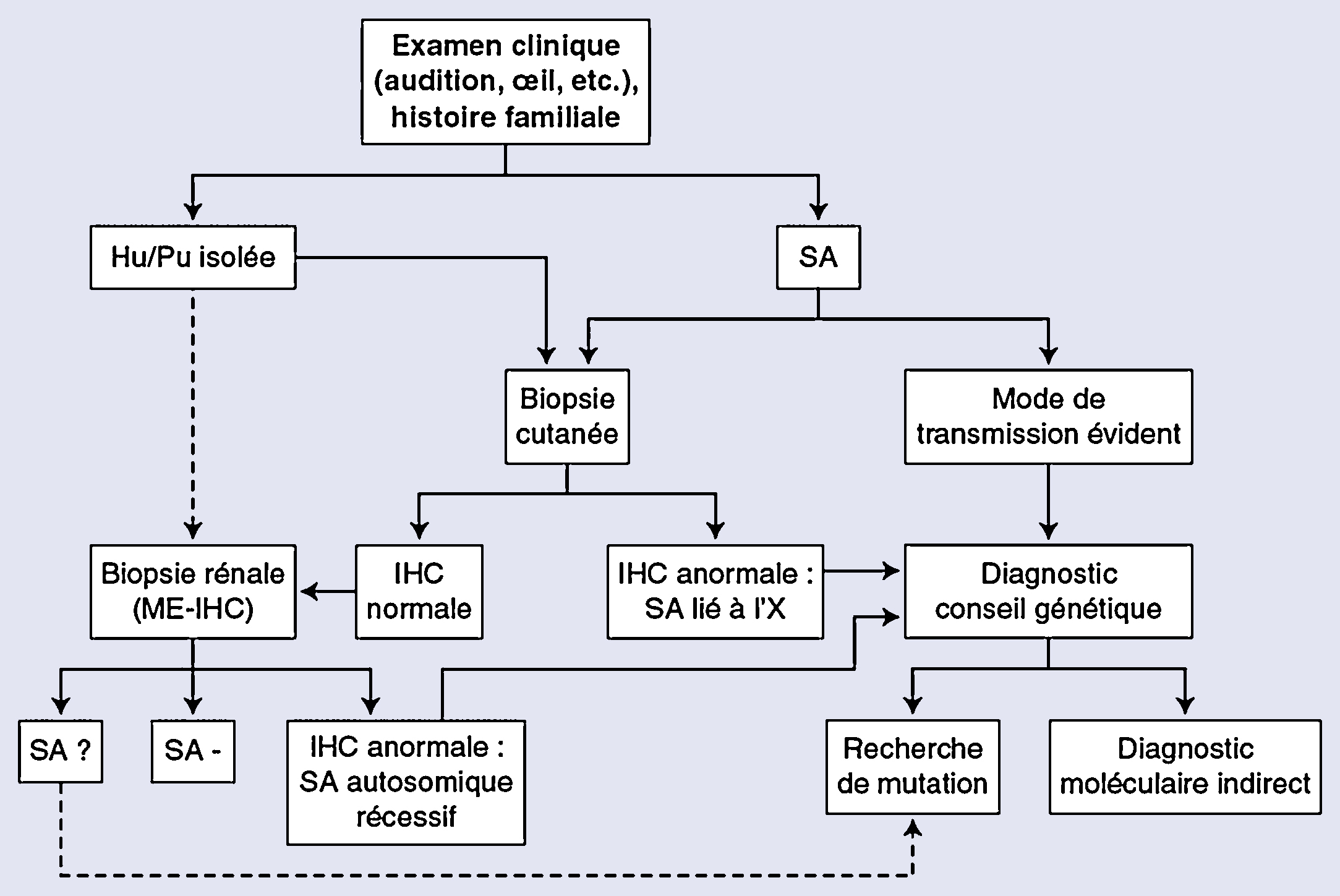
Le syndrome d’Alport est une affection héréditaire caractérisée par l’association d’une néphropathie hématurique progressive avec anomalies ultrastructurales des membranes basales glomérulaires, d’une surdité de perception d’évolution également progressive et parfois d’anomalies oculaires. Son incidence est de moins de 1 pour 5000 individus et cette maladie serait à l’origine d’environ 2 % des insuffisances rénales terminales en Europe et aux États-Unis. Le syndrome d’Alport est hétérogène d’un point de vue clinique et génétique. C’est une maladie du collagène IV, principal constituant des membranes basales, liée à des mutations dans les gènes codant l’une des trois chaînes, α3, α4 ou α5(IV), exprimées dans la membrane basale glomérulaire. Les mutations de COL4A5 sont associées au syndrome d’Alport lié à l’X, qui représente 80 à 85 % des cas et est plus sévère chez le garçon que chez la fille. Les mutations de COL4A3 ou COL4A4 sont associées aux formes autosomiques. L’étude de l’expression des chaînes de collagène dans les membranes basales cutanées et rénales permet de faire le diagnostic et d’établir le mode de transmission chez la majorité des patients. Il est nécessaire de savoir reconnaître ce syndrome étant donné son caractère familial, sa sévérité, et l’importance du conseil génétique. Les traitements bloqueurs du système rénine-angiotensine sont de plus en plus prescrits lorsqu’il existe une protéinurie. Des études prospectives seront nécessaires pour juger de l’efficacité de ces traitements sur la protéinurie et la progression de l’insuffisance rénale, et pour préciser les indications. Des études chez l’animal ont montré l’intérêt potentiel de différentes molécules (antiprotéases, bloqueurs des récepteurs aux chémokines, des inhibiteurs du transforming growth factor-β1, de l’hydroxy-méthyl-glutaryl-coenzyme A réductase, bone morphogenetic protein-7), des cellules souches hématopoïétiques et d’un anti-micro-ARN.
Alport syndrome is an inherited disorder characterized by the association of a progressive haematuric nephropathy with ultrastructural abnormalities of the glomerular basement membranes, a progressive sensorineural hearing loss and sometimes ocular involvement. Its incidence is less than 1 per 5000 individuals and the disease is the cause of about 2% of end stage renal disease in Europe and the United States. Alport syndrome is clinically and genetically heterogeneous. It is related to mutations in the genes encoding one of three chains, α3, α4 α5 of type IV collagen, the main component of basement membranes, expressed in the glomerular basement membrane. COL4A5 mutations are associated with X-linked Alport syndrome, which represents 80 to 85% of cases and is more severe in boys than in girls. Mutations in COL4A3 or COL4A4 are associated with autosomal Alport syndrome. The expression of collagen chains in skin and kidney basement membranes allows for the diagnosis and characterization of the mode of transmission in most patients. It is necessary to diagnose this syndrome because its family involvement, its severity, and the importance of genetic counseling. Angiotensin blockers are increasingly prescribed in proteinuric patients. Prospective studies are needed to assess the effectiveness of these treatments on proteinuria and progression of kidney failure, and to specify indications. Animal studies have shown the potential value of different molecules (protease inhibitors, chemokine receptor blockers, transforming growth factor-β1 inhibitors, hydroxy-methyl-coenzyme A reductase inhibitors, bone morphogenetic protein-7 inhibitors), hematopoietic stem cells, and of a anti-micro-RNA.