Néphrologie & Thérapeutique
MENUFamilial juvenile hyperuricemic nephropathy Volume 8, issue 2, Avril 2012

- Key words: Familial juvenile hyperuricemic nephropathy, Interstitial nephritis, Goutte precoce, Autosomal dominant disease, Uromoduline, Tamm-Horsfall protein
- DOI : 10.1016/j.nephro.2011.11.012
- Page(s) : 117-25
- Published in: 2012
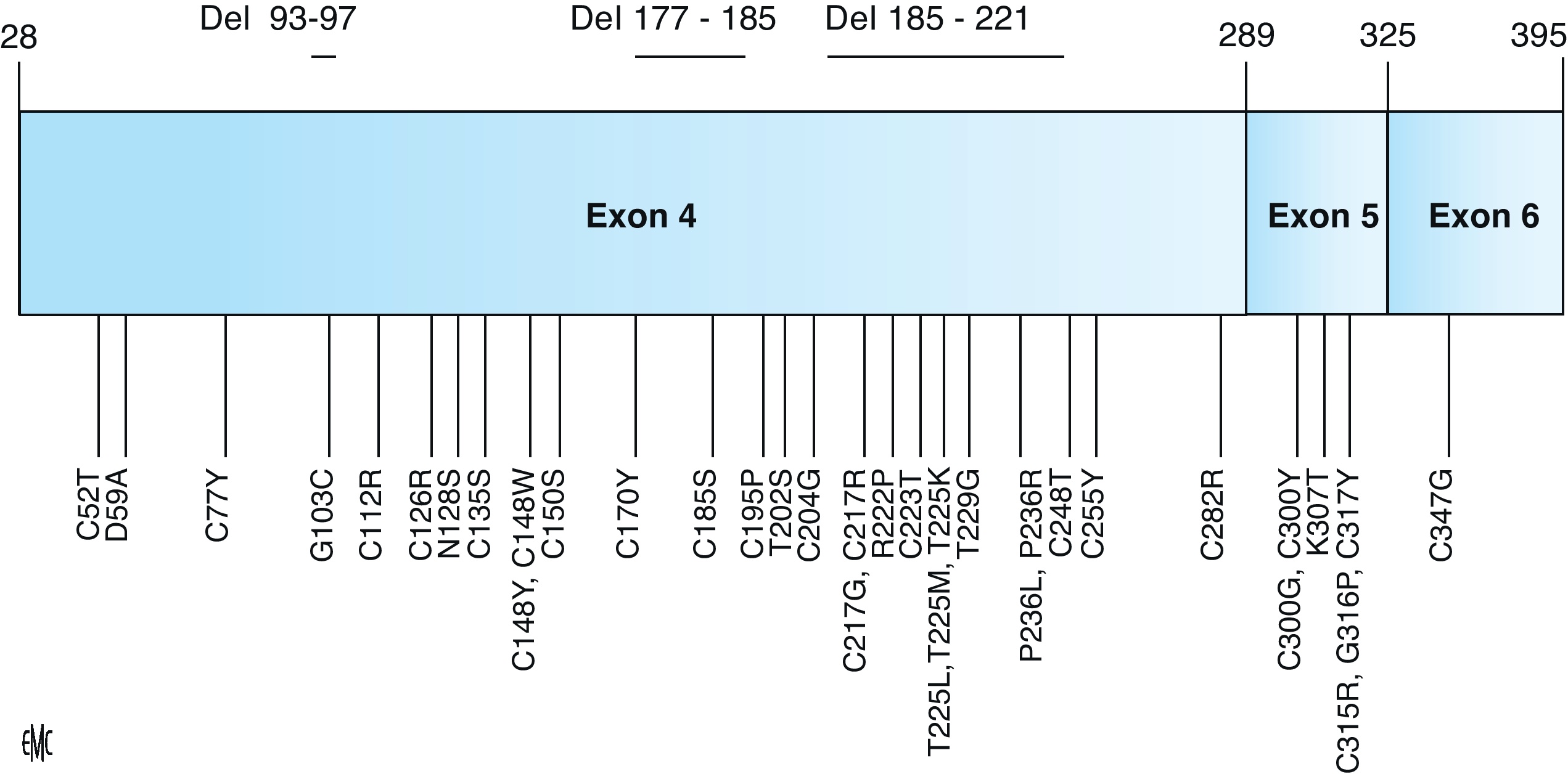
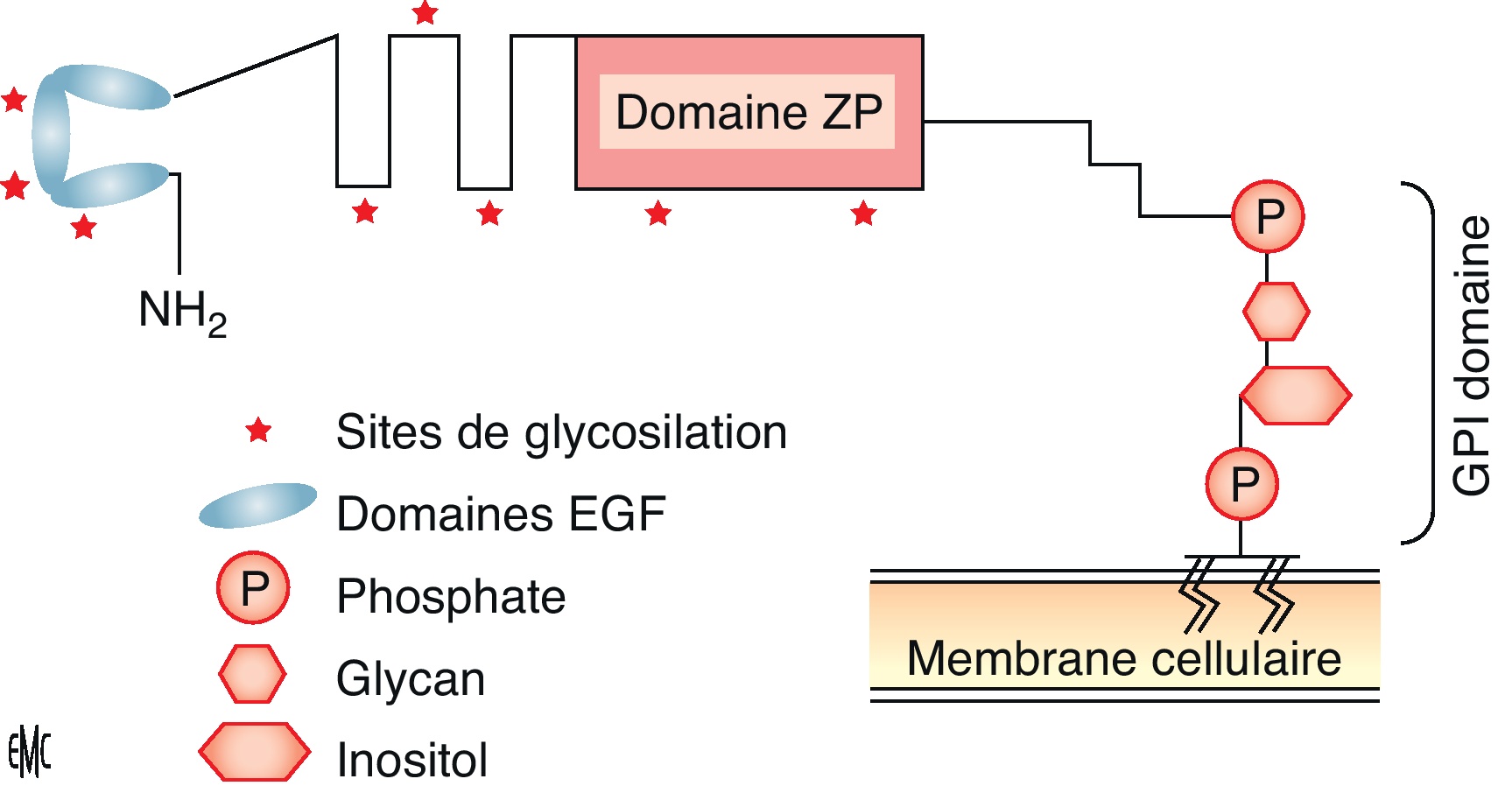
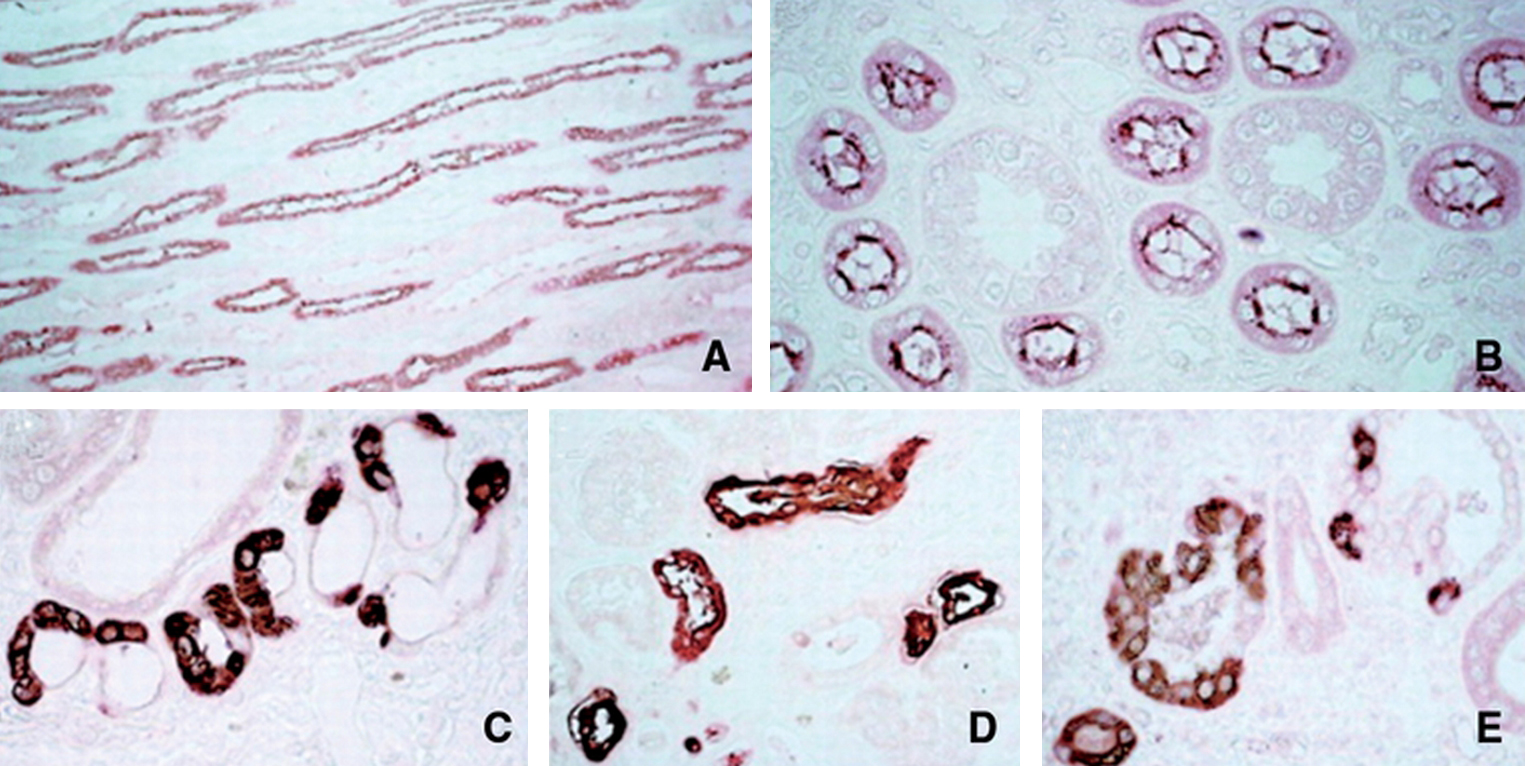
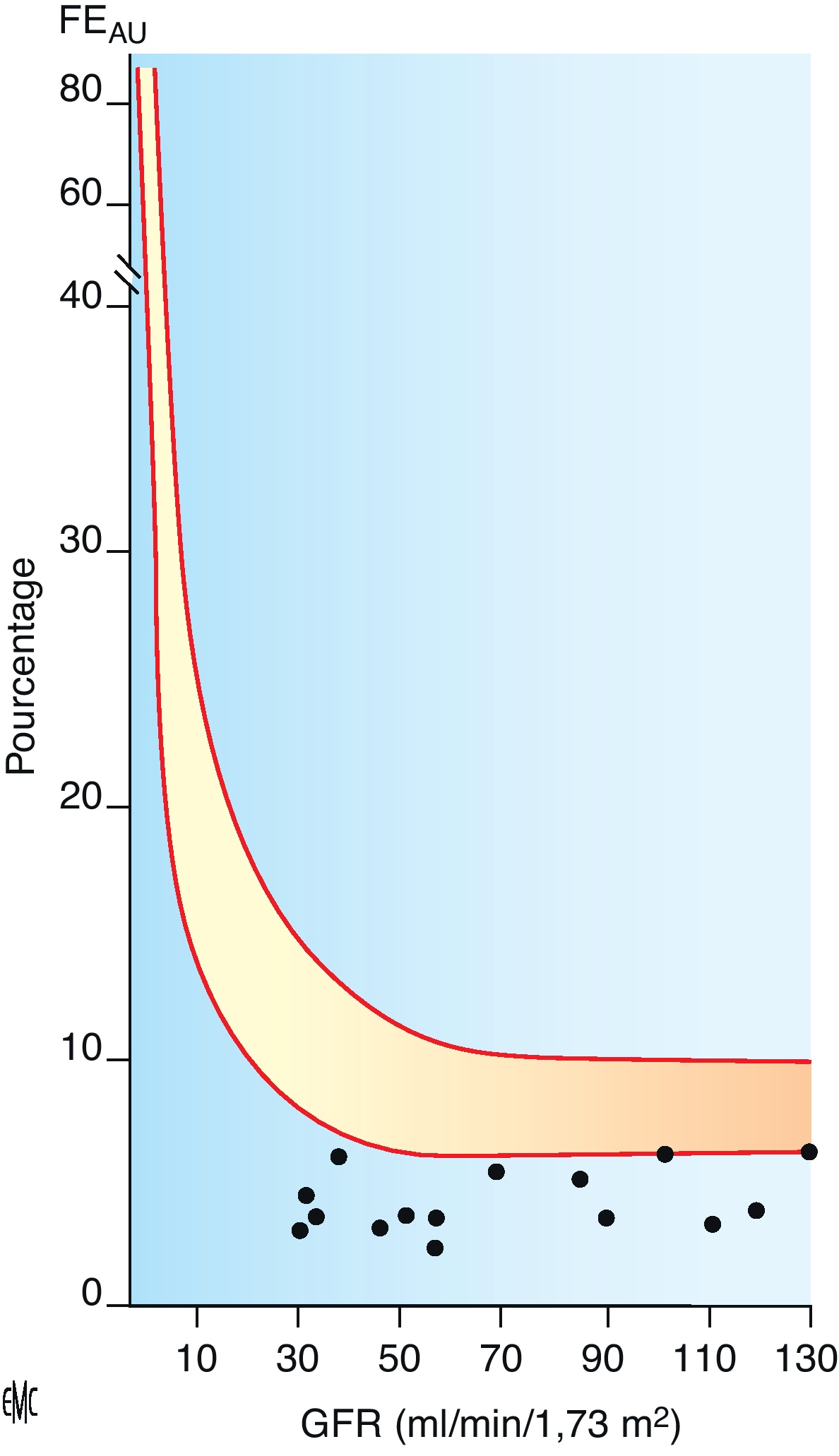
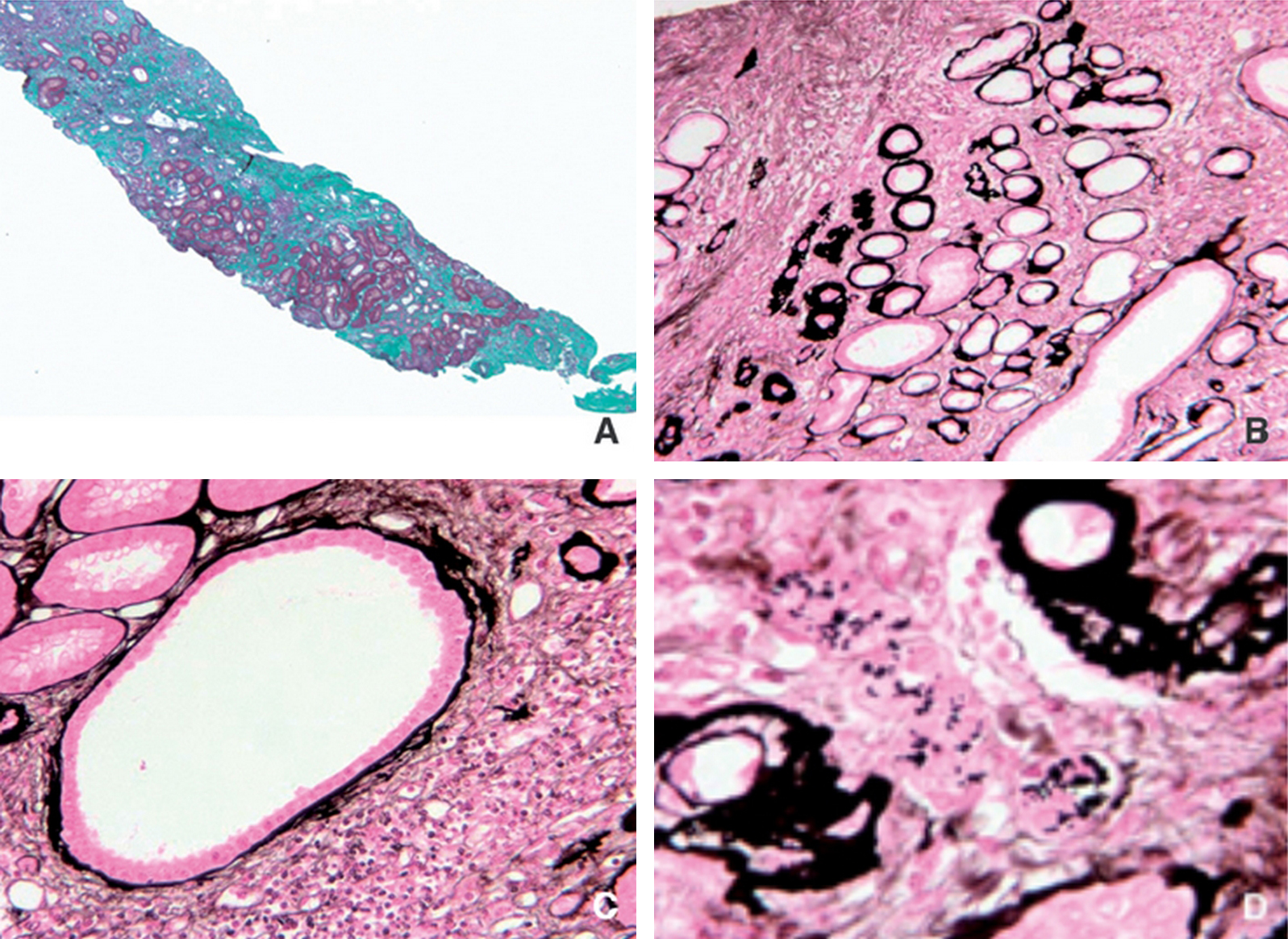
La néphropathie hyperuricémique familiale juvénile est une pathologie héréditaire rare se transmettant sur le mode autosomique dominant. Elle se caractérise par un défaut d’excrétion des urates responsable d’une hyperuricémie souvent compliquée de goutte précoce. L’atteinte rénale est une néphropathie tubulo-interstitielle. L’imagerie montre parfois des kystes en taille et nombre variables. La biopsie rénale bien que peu spécifique met en évidence une fibrose interstitielle parfois inflammatoire, des îlots de tubes atrophiques parfois dilatés et aux parois irrégulièrement festonnées. L’insuffisance rénale évolue progressivement vers le stade terminal entre 30 et 60ans. Le traitement par allopurinol est recommandé dès les stades précoces de la maladie, son efficacité sur le ralentissement de la maladie est incertaine. Il existe une hétérogénéité génétique dans cette maladie. Seul le gène uromoduline codant pour la protéine de Tamm-Horsfall a jusqu’ici pu être mis en cause dans moins de la moitié des familles. Les mutations décrites touchent le plus souvent une cystéine au sein de l’exon 4 entraînant une rétention de la protéine anormale dans le reticulum endoplasmique des cellules de l’anse de Henlé et une diminution de son excrétion urinaire. La physiopathologie de la maladie est encore incertaine. Les fonctions de la protéine de Tamm-Horsfall sont mal connues (rôles anti-infectieux, antilithiasique, immunomodulateur). Les souris knock-out ne développent pas de phénotype rénal mais sont plus sujettes aux infections urinaires à E. Coli. Des mutations du gène de l’uromoduline ont également été mises en évidence dans la maladie kystique de la médullaire, néphropathie tubulo-interstitielle autosomique dominante longtemps distinguée de la néphropathie hyperuricémique familiale juvénile. La génétique a permis de considérer ces deux entités comme les facettes d’une même maladie qu’il conviendrait d’appeler néphropathie associée à l’uromoduline. Au moins deux autres gènes ont été impliqués dans des tableaux cliniques similaires : TCF2 et le gène codant pour la rénine.
Familial juvenile hyperuricemic nephropathy is a rare autosomal dominant disease. It is characterized by abnormal handling of urate responsible for hyperuricaemia often complicated of gouty arthritis. Renal failure is due to tubulointerstitial nephritis. Ultrasonography sometimes finds renal cysts of variable size and number. Renal histology, although not specific, shows interstitial fibrosis, atrophic tubules, sometimes enlarged and with irregular membrane thickening. Renal failure progresses to end stage between 30 and 60 years of age. Allopurinol treatment is recommended at the early stages of the disease, its efficacy on slowing down the progression of the disease is however not proven. There is genetic heterogeneity in familial juvenile hyperuricemic nephropathy. Uromodulin encoding Tamm-Horsfall protein is the only gene to date identified, responsible in less than half of the families. The described mutations most often concern a cystein and are clustering in exon 4. These mutations result in abnormal retention of the protein in endoplasmic reticulum of Henle loop cells and in reduction of its urinary excretion. The pathophysiology of the disease is however still dubious. Indeed, Tamm-Horsfall protein functions are not well known (anti-infectious role, cristallisation inhibition, immunomodulating role). Knock-out mice do not develop renal phenotype but are more prone to E. coli urinary infections. Uromodulin gene mutations have also been described in medullary cystic kidney disease, an autosomal dominant tubulointerstitial nephropathy, considered at first as a distinct disorder. Genetic progress allowed us to consider familial juvenile hyperuricemic nephropathy and medullary cystic kidney disease as the two facets of a same disease, we should call uromodulin associated kidney diseases. At least two other genes have been implicated in similar clinical presentation: TCF2 and the gene encoding renin.