Néphrologie & Thérapeutique
MENUPrimary hyperoxaluria: A review Volume 12, issue 6, Novembre 2016

- Key words: Nephrocalcinosis, Oxalate, Primary hyperoxaluria, Urolithiasis
- DOI : 10.1016/j.nephro.2016.03.005
- Page(s) : 431-6
- Published in: 2016
Despite improvement in the quality of critical care, the incidence and mortality of acute kidney injury (AKI) continues to rise. The aim of our study was to analyze the changes during a 12-year period in etiology, incidence and outcomes of severe AKI, which required dialysis, in a large single centre.
We performed retrospective analysis of all the patients (n=3215) with severe AKI hospitalized and dialysed in the hospital of Lithuanian university of health sciences Kauno Klinikos (HLUHS KK) during the period of 2001–2012.
During a 12-year period, the incidence of severe AKI increased from 154 to 597 cases/p.m.p. The mean age of the patients increased from 58.2±19.2 years in 2001 to 65.7±17 years in 2012 (P<0.001). The number of men (n=2012; 62.6%) was significantly higher than that of women (n=1201; 37.4%; P<0.001). The causes of severe AKI were renal (n=1128; 35.1%), prerenal (n=642; 20%), obstructive (n=310; 9.6%) and in 12.7% of the patients-multifactorial. Overall, the most frequent cause of AKI was acute tubular necrosis (n=1069; 33.2%). The renal replacement therapy (RRT) was discontinued due to improved kidney function in 45.3% of cases. 8.1% of the patients remained dialysis dependent. The mortality rate was 44%.
During a 12-year period, the number of the patients with severe AKI increased three times with the predominance of men and elderly people. There was an observed increase in multifactorial causes of severe AKI; however, ATN remained dominant over the decade. The mortality rate remained high, almost half of the patients died, less than 10% remained dialysis dependent, the rest had the improvement of renal function.
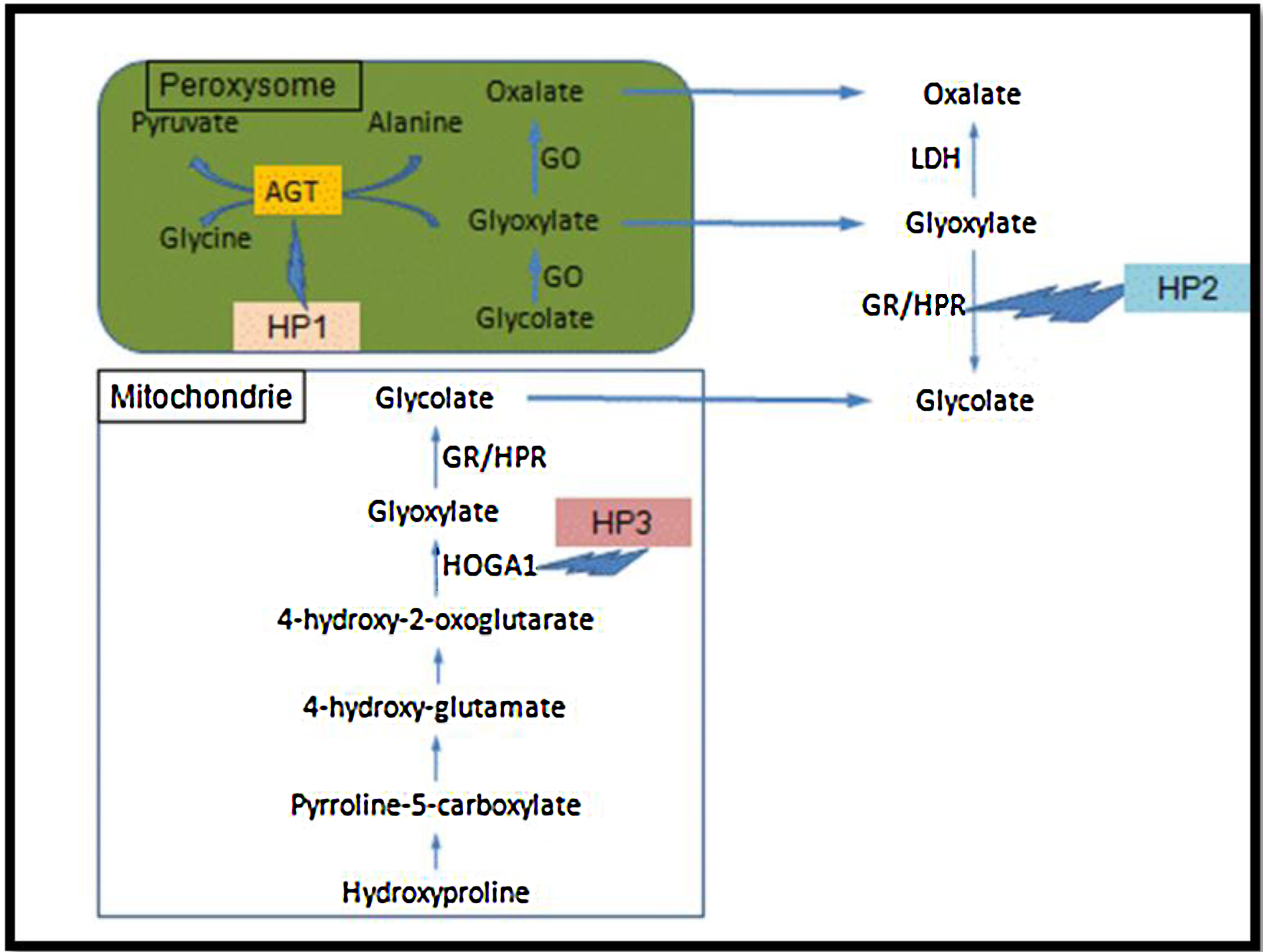
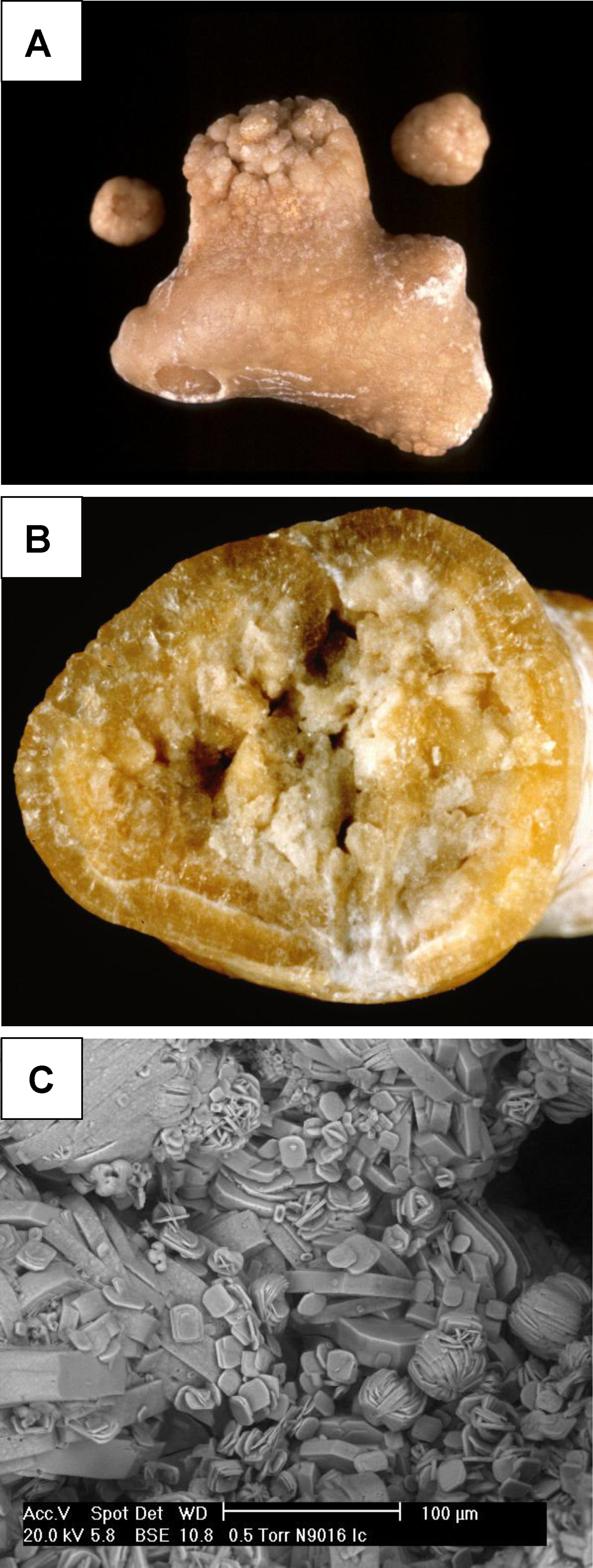
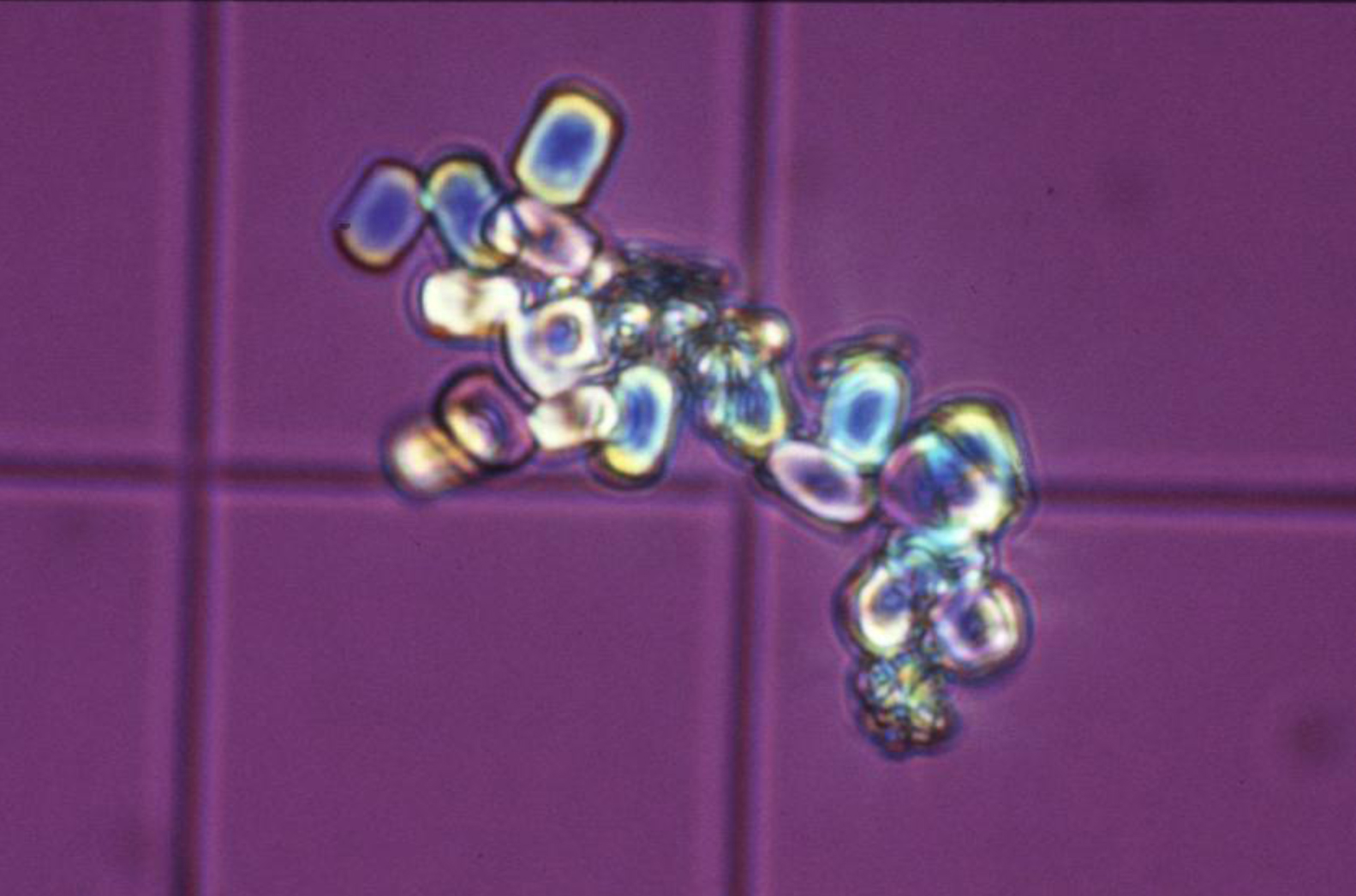
Les hyperoxaluries primaires (HP) sont des maladies liées à des erreurs innées du métabolisme du glyoxylate et de l’oxalate, transmises selon le mode autosomique récessif. Cette pathologie entraîne une augmentation de la production endogène de l’oxalate qui se traduit par une hyperoxalurie. L’HP de type 1, la plus fréquente, est due à un déficit d’une enzyme peroxysomale hépatique spécifique : l’alanine glyoxylate aminotransférase (AGT). L’HP de type 2 est secondaire à un déficit de la glyoxylate réductase/hydroxypyruvate réductase présente au niveau du cytosol des hépatocytes et des leucocytes. L’HP de type 3 est liée au gène HOGA1 codant pour une enzyme mitochondriale, la 4-hydroxy-2-oxo-glutarate aldolase. Les lithiases récidivantes et la néphrocalcinose constituent les marqueurs de cette maladie et conduisent à une dégradation progressive de la fonction rénale. Lorsque celle-ci est sévèrement altérée, l’augmentation des oxalates sanguins est responsable d’une oxalose systémique, c’est-à-dire l’accumulation d’oxalate de calcium dans les os, les viscères et l’appareil circulatoire. Le diagnostic, souvent tardif, est basé sur l’analyse du calcul, l’étude de la cristallurie, le dosage de l’oxalurie et l’analyse de l’ADN. Une prise en charge thérapeutique précoce basée sur l’hyperhydratation, les inhibiteurs de la cristallisation et l’administration de la pyridoxine (dans l’HP de type 1), pourrait ralentir la progression vers l’insuffisance rénale terminale qui, à ce stade, nécessitera une transplantation hépato-rénale qui pourrait corriger le déficit enzymatique.
Primary hyperoxalurias (PH) are inborn errors in the metabolism of glyoxalate and oxalate with recessive autosomal transmission. As a result, an increased endogenous production of oxalate leads to exessive urinary oxalate excretion. PH type 1, the most common form, is due to a deficiency of the peroxisomal enzyme alanine: Glyoxylate aminotransferase (AGT) in the liver. PH type 2 is due to the deficiency of the glyoxylate reductase/hydroxypyruvate réductase, present in the cytosol of hepatocytes and leucocytes. PH type 3 is linked to the gene HOGA1, encoding a mitochondrial enzyme, the 4-hydroxy-2-oxo-glutarate aldolase. Recurrent urolithiaisis and nephrocalcinosis are the markers of the disease. As a result, a progressive dysfunction of the kidneys is commonly observed. At the stage of severe chronic kidney disease, plasma oxalate increase leads to a systemic oxalosis. Diagnostic is often delayed and it based on stone analysis, cristalluria, oxaluria determination and DNA analysis. Early initiation of conservative treatment including high fluid intake and long-term co-administration of inhibitors of calcium oxalate crystallization and pyridoxine, could efficiently prevent end stage renal disease. In end stage renal failure, a combined liver–kidney transplantation corrects the enzyme defect.