Néphrologie & Thérapeutique
MENUPrimary hyperoxaluria Volume 7, issue 4, Juillet 2011

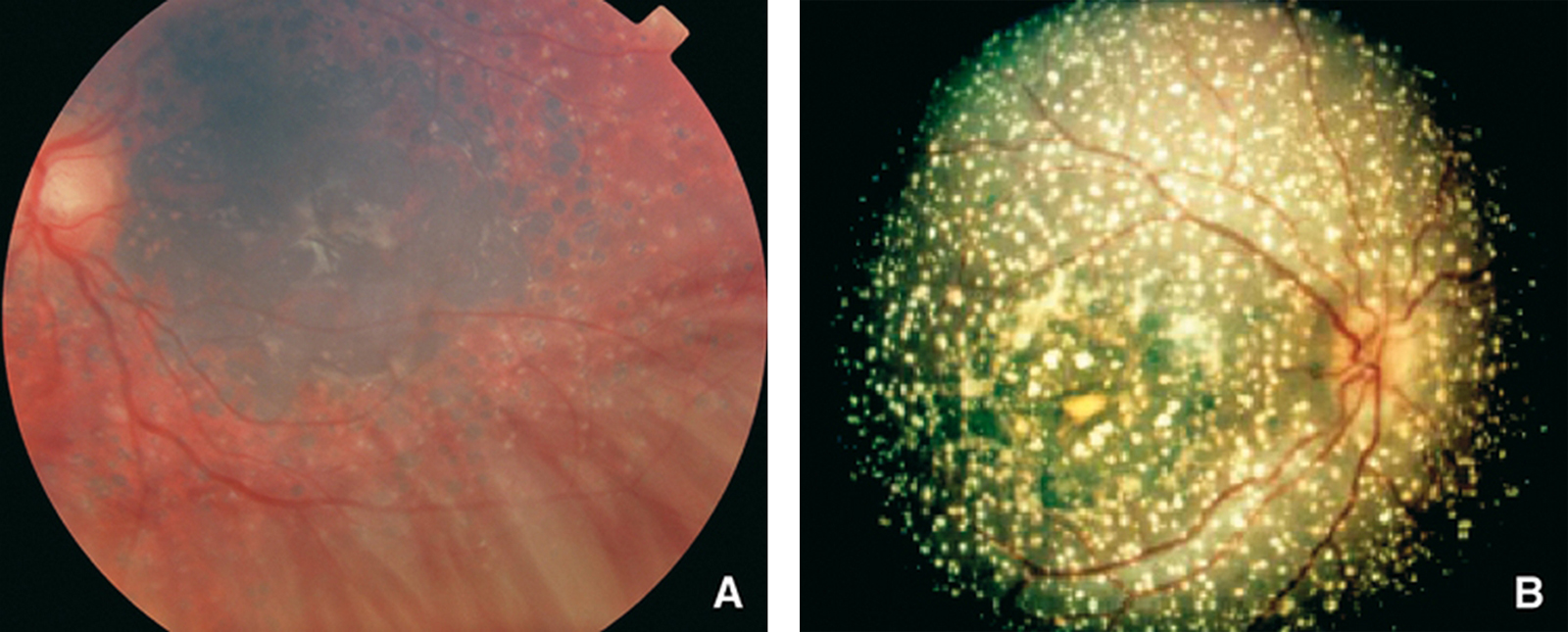
Figures

Tables
- Key words: Oxalosis, Primary hyperoxaluria, Nephrolithiasis, Nephrocalcinosis, Pyridoxin, Combined liver and kidney transplantation
- DOI : 10.1016/j.nephro.2011.03.004
- Page(s) : 249-59
- Published in: 2011
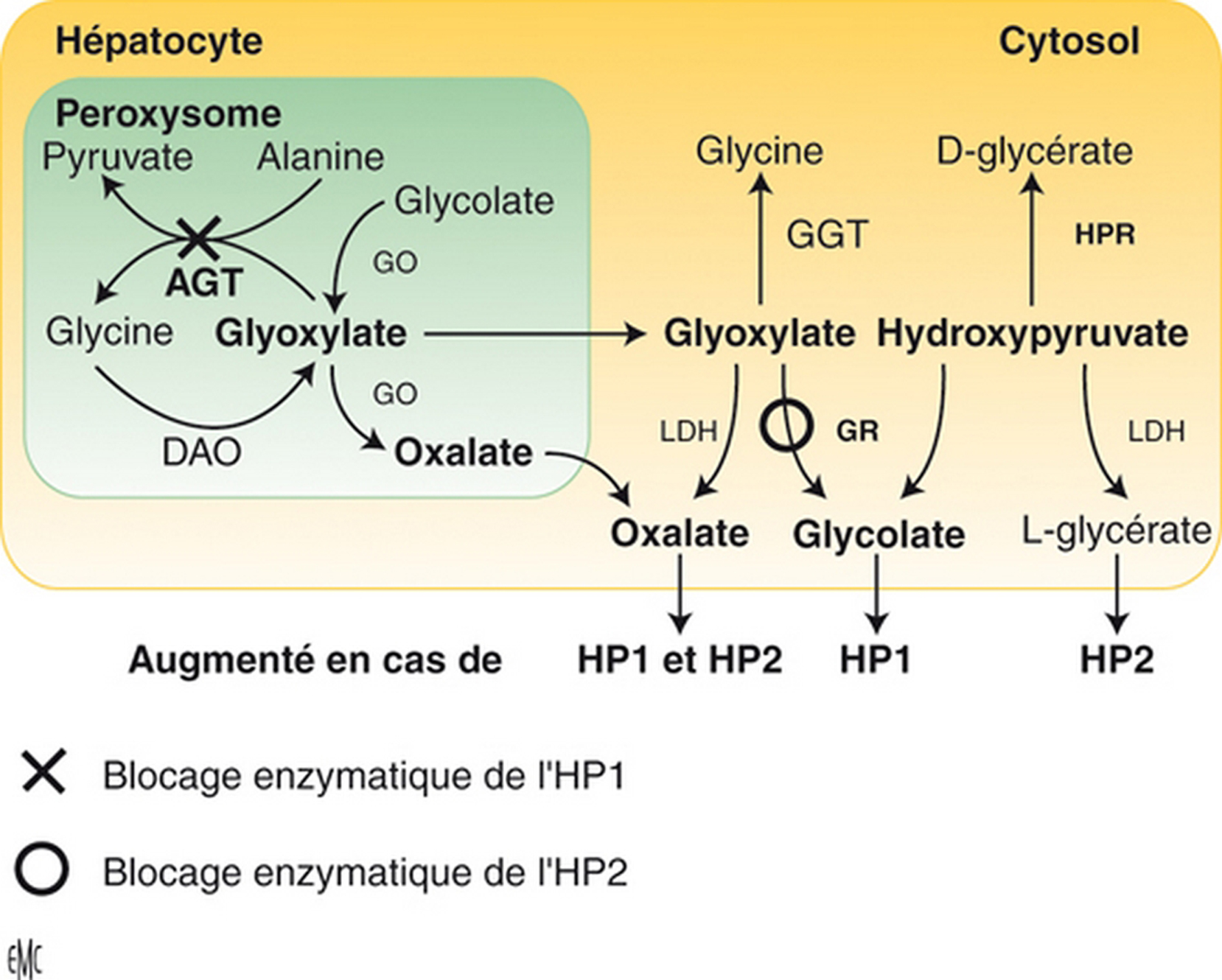
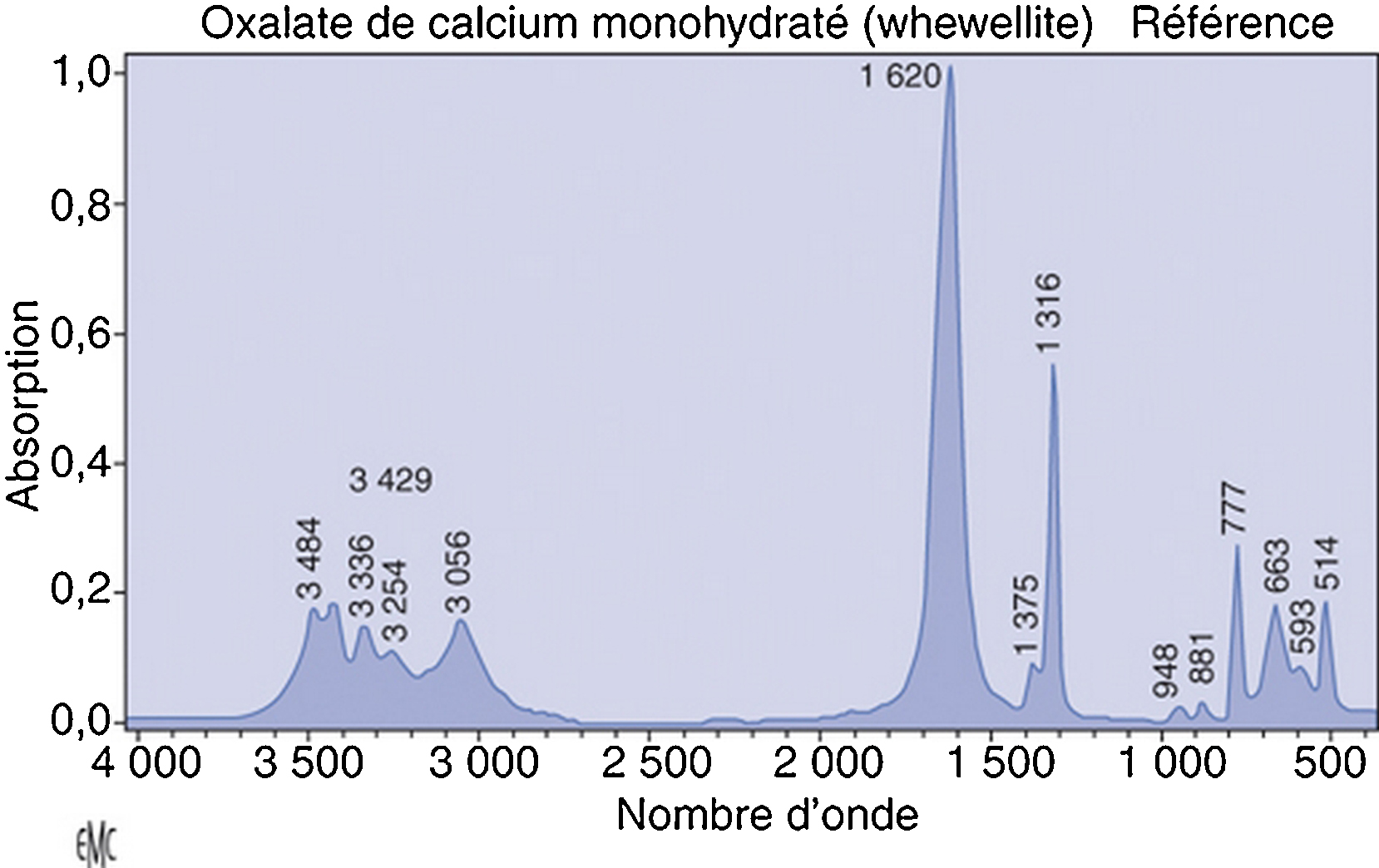
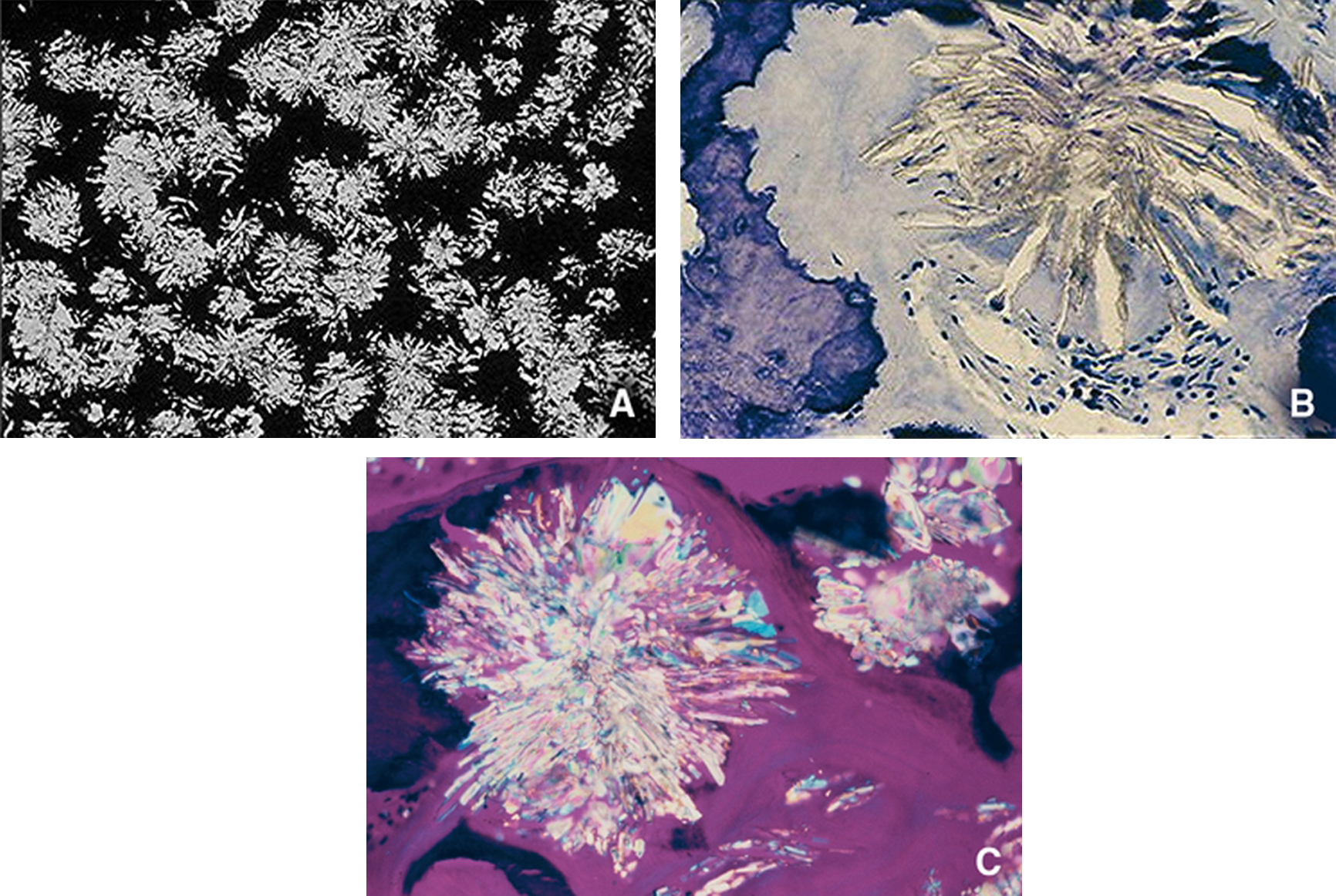
Les hyperoxaluries primitives sont transmises sur le mode autosomique récessif ; il s’agit d’affections aussi rares que graves, engageant toujours le pronostic rénal et parfois aussi le pronostic vital, notamment dans les formes à début précoce. Le type 1, de loin le plus fréquent en Europe, résulte d’un déficit enzymatique (alanine-glyoxylate aminotransférase) au niveau des peroxysomes du foie, à l’origine d’une hyperoxalurie qui s’exprime initialement par des lithiases avec ou sans néphrocalcinose. Au fur et à mesure que la filtration glomérulaire se dégrade, une surcharge systémique apparaît et n’épargne aucun organe, mais l’essentiel du stockage de l’oxalate se fait au niveau du squelette. Le diagnostic repose sur l’hyperoxalurie et la confirmation du type d’hyperoxalurie est le plus souvent établie par l’analyse moléculaire (qui permet aussi un diagnostic prénatal). Le traitement conservateur (hyperhydratation, inhibiteurs de la cristallisation, pyridoxine) est essentiel et doit être entrepris le plus précocement possible. Aucune méthode de dialyse n’est suffisamment efficace pour compenser la surproduction d’oxalate, de sorte que la transplantation hépatique et rénale doit être planifiée relativement tôt, avant le stade d’insuffisance rénale avancée, pour limiter les dégâts de la thésaurismose. Il se peut que, à l’avenir, de nouvelles thérapeutiques remplacent ou complètent la transplantation d’organes (transplantation d’hépatocytes, molécules chaperonnes, etc.).
Primary hyperoxalurias are rare recessive inherited inborn errors of glyoxylate metabolism. They are responsible for progressive renal involvement, which further lead to systemic oxalate deposition, which can even occur in infants. Primary hyperoxaluria type 1 is the most common form in Europe and is due to alanine-glyoxylate aminostransferase deficiency, a hepatic peroxisomal pyridoxin-dependent enzyme. Therefore primary hyperoxaluria type 1 is responsible for hyperoxaluria leading to aggressive stone formation and nephrocalcinosis. As glomerular filtration rate decreases, systemic oxalate storage occurs throughout all the body, and mainly in the skeleton. The diagnosis is first based on urine oxalate measurement, then on genotyping, which may also allow prenatal diagnosis to be proposed. Conservative measures – including hydration, crystallization inhibitors and pyridoxine – are safe and may allow long lasting renal survival, provided it is given as soon as the diagnosis has been even suspected. No dialysis procedure can remove enough oxalate to compensate oxalate overproduction from the sick liver, therefore a combined liver and kidney transplantation should be planned before advanced renal disease has occurred, in order to limit/avoid systemic oxalate deposition. In the future, primary hyperoxaluria type 1 may benefit from hepatocyte transplantation, chaperone molecules, etc.