Néphrologie & Thérapeutique
MENUBalancing the genetic risk of APOL1 kidney disease variants Volume 15, supplement 1, Avril 2019

- Key words: APOL1, Kidney disease, Selection, Trypanosome, Kidney transplantation
- DOI : 10.1016/j.nephro.2019.03.007
- Page(s) : 79-84
- Published in: 2019
Patients with end-stage renal disease that require chronic haemodialysis need a reliable vascular access. Unanimously, native arteriovenous fistulae are considered to be the most reliable access for patients with reasonable life expectancy. For the last 60 years arteriovenous fistulae have been created surgically at the wrist or the elbow with variable rates of success, maturation problems, reinterventions and complications, making this field of surgery particularly challenging and full of scientific controversies. The recent addition of the technical ability to create arteriovenous fistulae percutaneously comes to add one more option for the patients and one more source of controversy for the experts.
Necrotizing and crescentic rapidly progressive glomerulonephritis or crescentic glomerulonephritis is one of the severest forms of acquired glomerular diseases with significant mortality. Risk of end-stage renal failure at 5 years is near 30%, with a number of patients developing chronic kidney disease. Currently, autoimmune crescentic glomerulonephritides are treated with broad-spectrum immunosuppression inducing remission of the injury in the majority of patients. However, treatment is associated with significant side effects and by the time remission is achieved the majority of patients have developed renal tissue damage and significant impairment of their kidney function with a steep slope of deterioration within the first weeks following initiation of immunosuppression. It is therefore important to develop complementary strategies that would be immediately active on the common process of destructive epithelial processes. We have worked to identify the major cellular pathways contributing to glomerular destruction in this context by a systematic comparison of patient tissues and experimental models. Our studies demonstrate the pivotal role of local intra- and intercellular communications in orchestrating the global glomerular tolerance to a severe rapidly progressive glomerulonephritis model with excellent anatomoclinical correlative expressions in kidney biopsies of individuals diagnosed with crescentic glomerulonephritis, irrespectively of the causal immune disorder. We hope that such approaches deciphering mechanisms of cellular adaptation that underlie kidney damage control in response to vasculitides, integrating both stress and damage responses, will delineate novel complementary therapies.
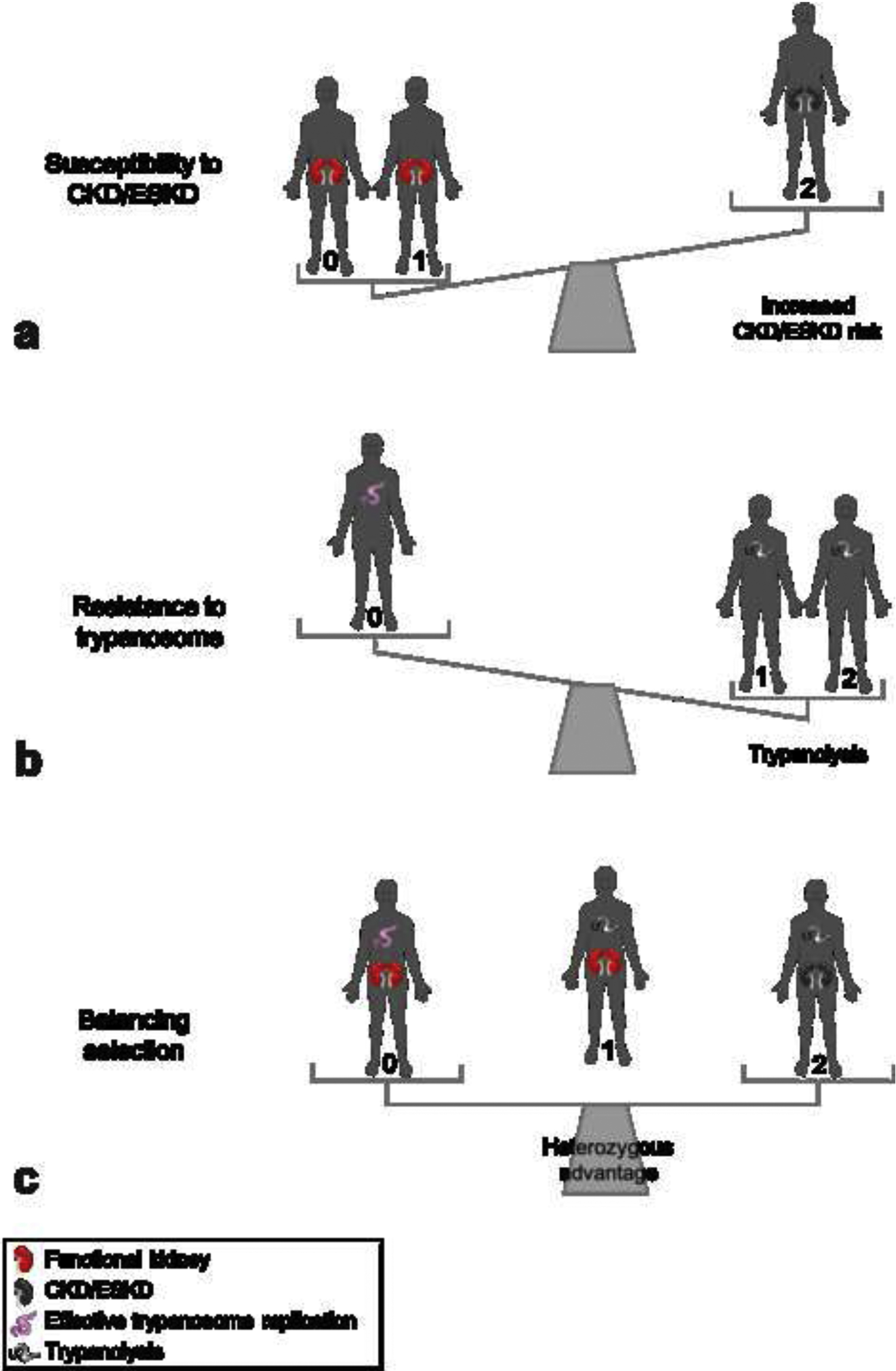
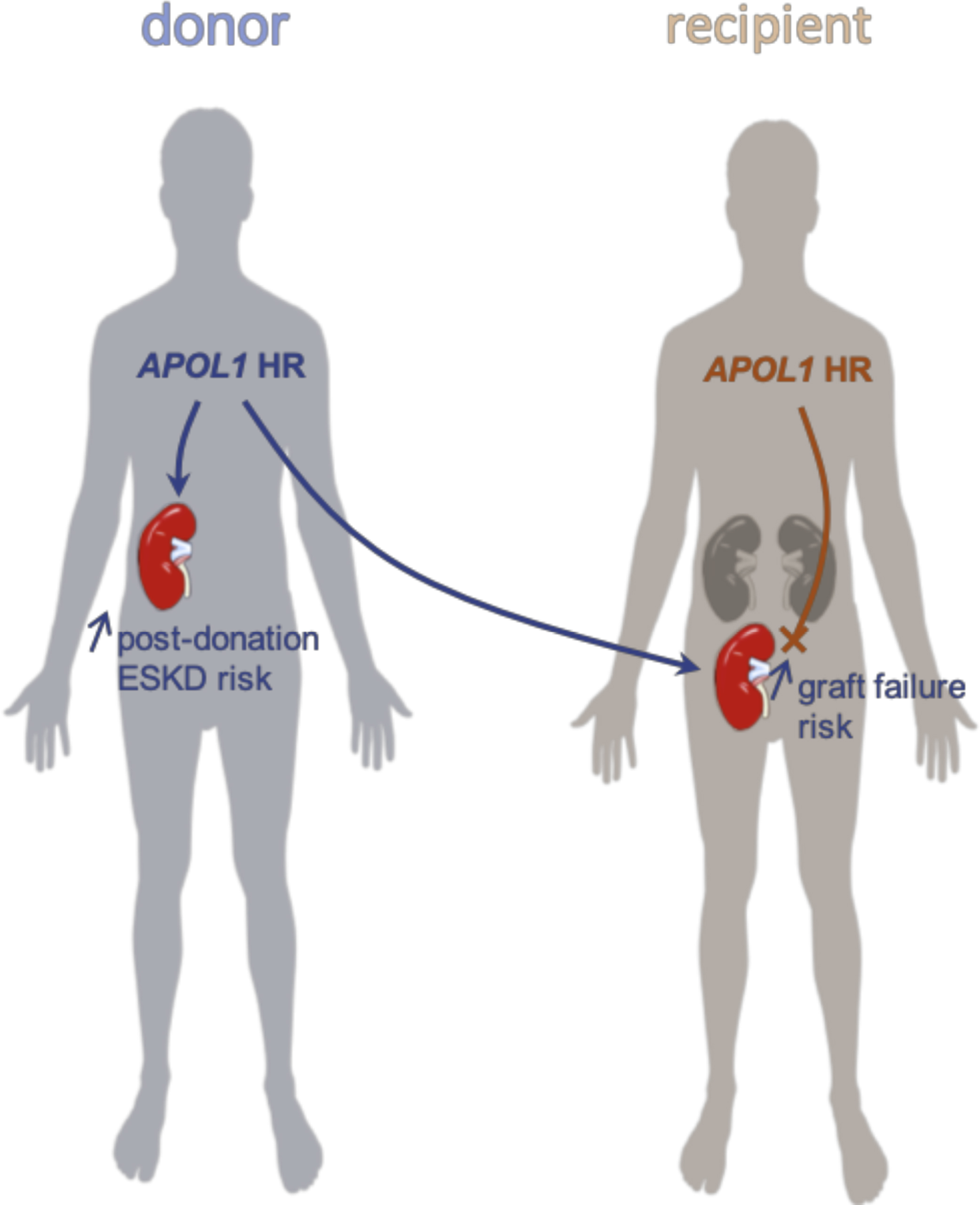
African-Americans exhibit an excess risk for chronic and end-stage kidney disease compared to the non-African populations. Two APOL1 genetic variants were shown to account for the majority of this racial disparity in glomerulopathies and other non-diabetic kidney disease. The high-risk genotype has only been reported in populations with recent African ancestry (14 % in African-Americans and up to more than 30 % in West Africa). In less than 10 years, the community has accumulated extensive knowledge on APOL1 and its genetic variants, from their positive selection for resistance against African trypanosomes to potential molecular mechanisms of podocyte injury. Finally, APOL1 associations with kidney transplantation outcomes and with postdonation end-stage kidney disease in living donors have paved the way for a personalized medicine implementation of APOL1 genotyping.