Hématologie
MENUWiskott-Aldrich syndrome Ahead of print

History
Wiskott-Aldrich syndrome (WAS) was discovered in 1937, when three brothers developed congenital thrombocytopenia, bloody diarrhoea, eczema and recurrent infections. Seventeen years later, Dr Aldrich demonstrated the X-linked nature of the condition [1].
This clinical description has been enriched over the years by certain immunological features with a better understanding of the associated immune deficiency: lymphopenia, defects in the production of polysaccharide antibodies and in the response to a variety of bacteria, and viral antigens. Similarly, platelet abnormalities are now better understood, as well as the clinical-biological description of the syndrome, which has been enriched by the increased risk of autoimmune diseases and cancers [2, 3].
WAS is an autosomal recessive X-linked inherited immune disorder (IID). It is caused by mutations in the Wiskott-Aldrich (WAS) gene located on the short arm of the X chromosome (Xp11.22-p11.23), encoding the WAS protein (WASp). Its incidence is estimated to be around one in 200,000 births. Despite X-linked inheritance, a few cases of girls with Wiskott-Aldrich syndrome have been reported in the literature, secondary to a defect in X-chromosome inactivation or double heterozygosity [3, 4]. The most severe forms occur very early in life, sometimes from birth, often initially with severe thrombocytopenia and then, very quickly, infections or autoimmune complications such as vasculitis. In addition to the classic severe phenotypic form of WAS, a form classically considered to be “attenuated”, known as X-linked hereditary thrombocytopenia (XLT), has also been described. Both diseases were initially linked to the Xp11 locus. Sequence analysis of the WAS gene identified distinct mutations responsible for the two phenotypes. The XLT form is now described as a variant of WAS, with less severe immune deficiency [3]. Nevertheless, it deserves special attention, as it can also be complicated, over time, by autoimmunity (vasculitis, autoimmune cytopenias, etc.) and cancers (mainly lymphomas) [5]. Reference is now made to patients with WAS/XLT, to discontinue the use of the obsolete term XLT. We will present the genetic description of this pathology, its physiopathology, its clinical-biological diagnosis, the differential diagnoses to be evoked as well as the possible symptomatic and curative treatments.
Genetic description
The WAS gene (OMIM#301000) was located by linkage analysis to Xp11.22-p11.23 and was sequenced in 1994 [2]. It consists of 12 exons containing 1,823 base pairs and encodes the WASp protein, which consists of 502 amino acids. WASp is expressed in the cytoplasm of all non-erythroid lineages derived from haematopoietic stem cells (HSC).Mutations resulting in a complete absence of WASp expression are most often associated with the classic WAS phenotype and an increased risk of developing autoimmune and/or oncological complications, whereas mutations resulting in residual expression of WASp are associated with the XLT phenotype, which is generally associated with lower morbidity and a better prognosis. Although there is no formal correlation between genotype and phenotype, these forms with reduced synthesis of the WASp protein are most often secondary to missense mutations located mainly in the first two exons of the WAS gene (76 %) [6].Female vectors have a 50% chance of passing the disease on to their sons. Prenatal diagnosis is feasible for male foetuses when the causative mutation has been identified in the family, but de novo mutations also occur [7].
Reverting forms
There is a high frequency of secondary genetic events leading to spontaneous reversion of the mutation in patients with WAS/XLT, with the appearance of cells re-expressing WASp, creating a somatic mosaic. These events may correct the pre-existing mutation or may consist of the appearance of a second mutation of the WAS gene restoring a reading frame. These revertant cells have been detected mainly among T cells and more rarely in B cells and natural killer (NK) cells, with a prevalence varying from 1 to 80% depending on the patient. These revertant events have been observed in individuals carrying certain mutations located in specific domains of the WAS gene, and the mechanisms underlying them remain unclear. Like other primary immunodeficiency disorders (PIDs) in which somatic mosaicism is sometimes observed (e.g. adenosine deaminase-deficient severe combined immune deficiency, ADA-SCID, X-SCID, recombination activating gene 1/2 deficiency, RAG1/2), WASp-expressing revertant cells are generated by random events and are subsequently selected for their survival and/or proliferative advantage. The higher number of revertant forms of WAS/XLT than in other PIDs may be related to the fact that many patients with WAS/XLT do not benefit from curative treatments and survive longer than other PIDs. The presence of revertant cells does not protect these patients from developing severe forms and complications [8].
In addition, germline “gain-of-function” mutations in WAS (activating mutations in the GDB domain) have been described in patients with profound central neutropenia associated with myelodysplasia of varying degrees [9].
This nosological entity, referred to as XLN (for X-linked neutropenia), is different from WAS/XLT and will not be described here.
Pathophysiology, score and clinical impact (figure 1)
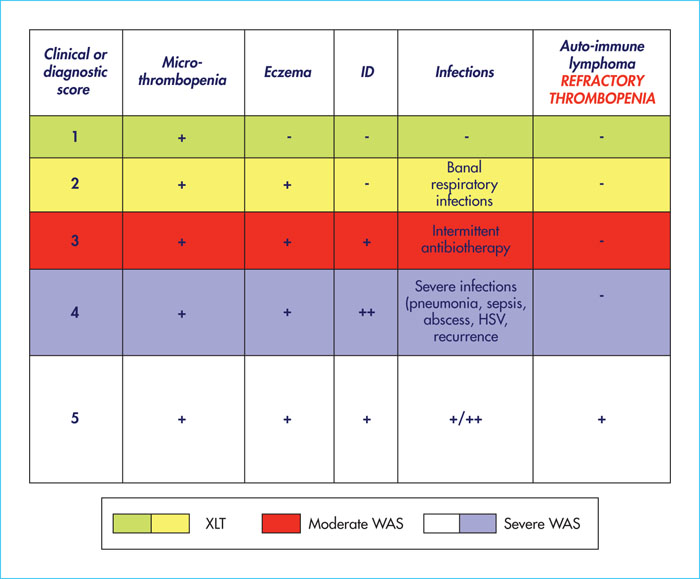
The clinical signs of WAS/XLT classically combine the following triad in boys: microthrombocytopenia, repeated severe or opportunistic infections (related to the immune deficiency), and eczema (of variable intensity). A disease severity score (figure 2) ranging from 1 to 5 is useful to assess the level of severity (from least to most severe) [6, 10]. This score is progressive, and an autoimmune disease or cancer can occur at any age, placing the patient in the highest severity score (score 5). Patients with a mild phenotype in childhood may develop autoimmune and onco-haematological manifestations (especially lymphoma) later in life (including adulthood).
Role of the WASp protein
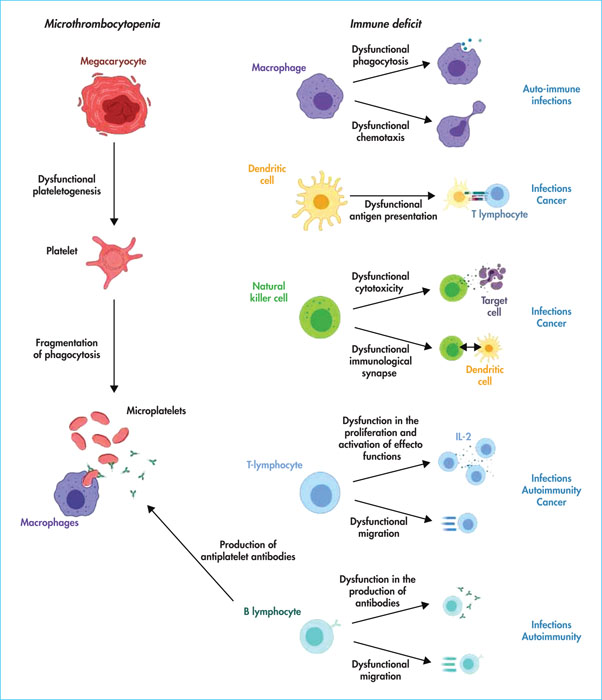
WASp is expressed in CD34+ haematopoietic progenitor cells and all lineages derived from them with the exception of erythroid lineages. All B cells, T cells, NK cells, monocytes and dendritic cells (DCs) in patients with WAS/XLT show abnormalities in signalling and cytoskeleton formation. In its basic state, the WASp protein is in its auto-inhibited form. Once activated by proteins of the Rho-GTPase (Ras homologous) family such as Cdc4215 (for cell division control protein 42), WIP (for WASp interacting protein), or phosphatidylinositol biphosphate (PIP2), WASp associates with the Arp2/3 (for actin related protein) complex. It thus initiates the polymerisation of G-actin into microfilaments (F-actin) [11]. WASp is thus a regulator of the actin cytoskeleton, providing a signal for actin filament polymerisation and rapid rearrangement of the actin cytoskeleton. This dynamic of the actin cytoskeleton is crucial for establishing processes such as cell mobility, phagocytosis and immunological synapse formation. All clinical symptomatology is related to the degree of abnormality of these functions [12].
Thrombocytopenia
There is a particularly severe form of thrombocytopenia in patients with WAS, especially infants, which is similar to a severe score 5 event. It is characterised by profound thrombocytopenia (< 10 G/L) refractory to all lines of treatment and for which an allogeneic HSC transplant (even in a haplo-identical situation) is urgently required, given the risk of haemorrhage [10].
The mechanisms leading to thrombocytopenia have long been debated and appear to be multifactorial. While megakaryocytes are normal or even increased in number in the bone marrow, with normal platelet production in vitro, studies have shown evidence of inefficient thrombopoiesis in vivo. Patients with WAS/XLT do not have increased levels of cross-linked platelets and consequently have low levels of immature platelet fractions, which encourages a defective central process, through a mechanism of abnormal plateletogenesis [12, 13].
However, there are also several pieces of evidence for peripheral destruction, accounting for the thrombocytopenia. Thus, in patients with WAS, abnormal platelet formation shows fragmentation of platelets in the periphery, responsible for microthrombocytopenia. However, the most convincing argument is that thrombocytopenia corrects itself in almost all cases after splenectomy, even if platelet abnormalities remain. There is also an increase in ex vivo phagocytosis of platelets from patients with WAS by macrophages compared to platelets from healthy subjects [14]. On the basis of these observations, it is therefore accepted that intrinsic platelet defects, including platelet membrane stiffness, in patients with WAS/XLT lead to increased phagocytosis by the reticuloendothelial system in the spleen and medulla, thereby reducing their survival. Importantly, anti-platelet antibodies are often detected in patients with WAS/XLT (60–70% of cases) and increase the severity of thrombocytopenia. Some studies have shown the existence of a thrombopathy consisting of a platelet aggregation defect [15, 16].
Immune deficiency
Patients with WAS/XLT are at risk of developing bacterial, viral and fungal infections, due to the impairment of all immune cell lineages with consequent impairment of humoral, cellular and innate immunity. The most common infections occur after the age of six months, due to the protection afforded by maternal immunoglobulin (Ig) G before this age. Bacterial infections are mainly lung infections, acute otitis media, septicaemia and meningitis, by encapsulated germs such as Streptococcus pneumoniae, Haemophilus influenzae type b and Staphylococcus aureus. There is also a susceptibility to herpes viral infections: Herpes simplex, varicella zoster virus, Epstein-Barr virus (EBV) and cytomegalovirus. Opportunistic infections, such as pneumocystis, have also been reported but are rarer. The fungal infections described (10% of cases) are of variable severity and usually caused by fungi such as Candida and Aspergillus[16].
Innate immunity
The innate immunity compartment of patients with WAS/XLT is altered with innate immunity cells, such as polymorphs, monocytes and other phagocytic cells, in normal amounts but with abnormal chemotactic function and diminished phagocytic capacity. Hypereosinophilia and elevated IgE levels may be observed. Some studies have also shown that the N-terminal part of WASp interacts with the SH3 domain of Btk. This interaction is crucial for TLR2 (Toll-like receptor 2) and TLR4 activation-induced signalling in macrophages, suggesting impaired initiation of the inflammatory response in patients with WAS/XLT [17]. NK cells are normal or increased in number, but their function is impaired due to a defect in the formation of the immunological synapse and their cytolytic function. Tumour immunosurveillance of T cells, but also of NK cells and DCs, seems to be impaired and could explain the occurrence of cancers. The mechanisms, which are still hypothetical, are thought to be a defect in the NK/DC dialogue associated with impaired migration of NK and DC cells and a dysfunction in DC phagocytosis [18].
Adaptive immunity
Cellular adaptive immunity is quantitatively and qualitatively impaired by:
- –a dysfunction in the presentation of antigens by DCs to T lymphocytes,
- –the regulatory role of WASp on the function of T and B lymphocytes, and thus in the response of cytotoxic T cells and the activity of T helper cells.
T lymphocytes
T lymphopenia is common and tends to involve naive and CD8 T-cells. The decrease in the number of naive T cells early in life could suggest a defect in thymopoiesis. Nevertheless, the overall lymphocyte count is >1000/mm3 in 78% of cases but is associated with a lack of lymphocyte response to mitogens in almost 50% of cases. During thymopoiesis, in mice lacking WASp (knock-out mice), it has been shown that thymic maturation is suboptimal, before the so-called double-positive cell stage, suggesting a role for WASp in thymic maturation. A tendency for spontaneous apoptosis of thymocytes has also been described. Once mature, T cells in patients with WAS/XLT are unable to polymerise and organise their actin cytoskeleton in response to stimulation of their T cell receptor (TCR). This leads to abnormal T-cell migration, defective signalling with a partial defect in effector function and survival. T lymphopenia worsens with age and the TCR diversity abnormalities associated with the initial thymopoiesis defect become more pronounced [19].
B lymphocytes and antibody production
Classically, these patients have dysgammaglobulinaemia with defective IgM production, abnormal IgA production (with defective IgA glycosylation), and an inadequate antibody response to vaccines (especially unconjugated pneumococcal vaccines) related to impaired antipolysaccharide antibody production. Polysaccharide antibodies are frequently lowered (decreased anti-Streptococcus pneumoniae and anti-Haemophilus influenzae type b responses in 66% and 69% of children with WAS, respectively) [20-22].
The study of B-cell subpopulations shows a decrease in the number of immature B-cells in the bone marrow. The hypothesis is a defect in the migration of B cells, with a decrease in their medullary retention time. Transitional B cells normally migrate from the bone marrow to the spleen, where the affinity/avidity of their B cell receptor (BCR) is tested against self antigens. In patients, the total number of B cells in the periphery is lower in childhood, with the level correcting in adulthood. In contrast, the distribution of peripheral B cell subtypes remains altered, with an increase in adult transitional B cells in the blood and an increase in plasma B cell activating factor (BAFF) levels, but a decrease in naive and memory B cells in older adults. The generation of somatic hypermutations is decreased, suggesting a defect in secondary maturation of B cells in the germinal centres, explaining a restriction of the antibody repertoire with consequent humoral inefficiency [22].
The immune deficiency is therefore both cellular (mainly CD8 lymphopenia) and humoral.
Autoimmunity
There is an increased risk of autoimmune diseases and vasculitis (all organs can be affected, particularly the joints, colon, skin, kidneys and central nervous system). The risk factors are not well identified. The frequency of such complications can reach 70% in some series [10, 16, 20]. Autoimmune and autoinflammatory events can occur at any age and are likely to be severe, difficult to treat, and to have an impact on patient survival [23]. Patients with WAS/XLT should be monitored renally, as a number of cases of IgA nephropathy (occurring at any age) have been reported [16]. Furthermore, autoimmunity remains a complication after allogeneic marrow transplantation in cases of mixed chimerism [23, 24]. Early studies on autoimmunity in these patients focused on characterising quantitative and qualitative defects in regulatory T cells (Treg). They showed that patients with WAS can generate normal numbers of naive Treg cells (nTreg) with an impaired function. Regulatory B cells (Breg) are decreased in patients with WAS/XLT, but their precise role in autoimmunity has not been studied further. Moreover, the proportion of memory B cells is decreased in adults with WAS and autoimmunity with an altered proliferative capacity and a decrease in somatic hypermutations, which have already been described as being associated with an increased self-reactivity of B cells towards polysaccharides already present on the surface of red blood cells. These data suggest that the mechanisms of tolerant B cell selection may be defective in this disease, with an oligoclonality of autoreactive B cells in patients with WAS and consequently an increased frequency of autoreactive antibodies (e.g. antinuclear [ANA], anti-double-stranded DNA [anti-dsDNA] or anti-insulin antibodies). Since these defects do not exist in immature B cells in the marrow, these events would occur during peripheral maturation of B cells as transitional B cells mature into naive B cells. The increase in BAFF observed in patients with WAS also suggests an increased selection or survival of these autoreactive B cells, enhanced by the lack of nTreg efficacy [21, 22].
These abnormalities in B cell homeostasis, together with abnormalities in Treg and potentially Breg function, may partly explain the incidence of autoimmunity in patients with WAS, although the exact mechanisms responsible for the defect in the deletion or positive selection of these autoreactive B cells remain to be elucidated. Nevertheless, the analysis of these autoreactive Bs could be a biomarker for monitoring B cell tolerance in patients with WAS after transplantation or gene therapy, especially in individuals with mixed chimerism, where the risk of autoimmunity persists.
IgA glomerulopathy is one of the classic autoimmune complications in WAS/XLT, occurring in approximately 3–19% of cases [16, 20, 25]. It is secondary to a defect in IgA glycosylation with increased levels of IgG-IgA immune complexes deposited in the renal glomeruli.
Atopy
An eczematous rash is an important clinical feature of the original description of the disease and affects more than 80% of patients at certain times in their lives [10]. The characteristics of the lesions correspond to the typical clinical diagnostic criteria of atopic dermatitis and can significantly affect patients’ quality of life. Hypereosinophilia and high IgE levels are observed. The underlying mechanisms continue to be a defect in Tregs and preferential differentiation of TCD4 into T auxillary 2 (TH2). Allergic rhinitis, asthma and food allergies are also observed in these patients with a higher prevalence than in the general population [16, 20, 25].
Cancer
As with most immune deficiencies, there is a risk of developing a malignant complication. This places the patient in the highest severity score (score 5). The prevalence of malignancy is estimated to be between 13 and 22% with a mean age at onset of 9.5 years and an increased risk in patients with autoimmune disease [10-25]. XLT patients should have a lower prevalence (5.2%), but this remains to be confirmed [25]. The possibility of developing cancer is therefore a major concern for all patients with WAS/XLT, regardless of their score. Lymphomas, including Hodgkin's lymphoma, are the most common, but lymphoblastic leukaemias, myelodysplasias, myeloproliferative syndromes and other malignant diseases (e.g. seminoma, testicular carcinoma, glioma, etc.) have also been described [10-25]. Analysis of the national registry of the Centre de Référence des Déficits Immunitaires Héréditaires (Ceredih) showed that these malignant lymphoproliferative syndromes were aggressive mature B lymphomas, frequently associated with EBV, and had the particularity of being frequently associated with a central neurological location. Mutations located at splice sites were more frequently associated with an increased risk of developing such complications. An HSC allograft is a treatment option for these patients, even in partial remission [5]. These patients also have a propensity to develop benign lymphoid proliferation due to follicular hyperreactivity or Kaposi's sarcoma [5].
The increased occurrence of these cancers may be explained by a decrease in immunosurveillance by dendritic cells, T cells and NK cells, as described above.
Conclusion
The clinical signs in adults differ little from those in children. However, if the diagnosis has not previously been made or if the patient has not been transplanted in childhood, it is likely that the patient has a less severe form of haematological and/or cellular and humoral deficiency. For these patients, the risk of autoimmunity and cancers remains high, and these complications should be considered in the presence of chronic thrombocytopenia refractory to usual treatments or with signs of vasculitis or associated cancers.
Diagnosis
There is a classification of disease severity coded from 0 to 5 according to clinical criteria [6]. This classification has been modified with the description of a particularly severe phenotype in infants (age < 2 years) with severe refractory thrombocytopenia, sometimes associated with autoimmunity and/or vasculitis and/or onco-haematological complications (figure 2). These children are classified as score 5 equivalent [10]. However, even patients with WAS/XLT who have a moderate phenotype (< 2 on the classification) require prolonged monitoring throughout their lives, with the risk of progressing to a more severe phenotype (score 3 to 5) at any time.
The diagnosis of WAS should be suspected in any boy with thrombocytopenia, either alone or with eczema and/or repeated ENT and/or respiratory tract infections and/or autoimmunity and/or a diagnosis of cancer. In order to make the diagnosis, the tests listed in table 1 are necessary.
Blood count
Patients with WAS/XLT have thrombocytopenia to varying degrees, but it is present in 100% of cases. Measurement of mean platelet volume (MPV) often reveals microthrombocytopenia but this may be confounded by a concomitant peripheral autoimmune mechanism, which may lead to MPV within the normal range due to the coexistence of small platelets, typical of WAS/XLT, and large platelets, common in immunological thrombocytopenia. Platelet volume is sometimes difficult to measure, and cytologists should be asked to assess the volume on a smear or to perform an MPV curve by cytometry. Cross-linked platelets are reduced in quantity. Other abnormal blood count findings may include anaemia and neutropenia, which may indicate autoimmunity, as well as lymphopenia, particularly in younger patients, or hypereosinophilia.
Immunoassay
Flow cytometry analysis of lymphocyte subpopulations often shows CD8 lymphopenia, sometimes a low B cell count in children and a decreased percentage of memory B cells in adults.
Humoral immunity studies often report normal or increased IgG values, low levels of IgM and anti-A and anti-B allohemagglutinins, and high IgA levels. There may be increased IgE. Vaccine antibody titres may be low or normal, while responses to unconjugated pneumococcal antigens are usually defective.
Lymphocyte proliferations in response to mitogens, tetanus toxoid and candidin may show normal or low responses but are not necessary for diagnosis.
Expression of the WASp protein
Intracellular expression of WASp in peripheral blood leukocytes can be analysed by flow cytometry, allowing rapid diagnosis when the result shows a complete absence of WASp detection compared to a healthy control. However, residual WASp expression or the existence of two populations in case of reversion may make the interpretation of the results more difficult. This technique will therefore be performed as a second line of defence after genetic analysis, in order to provide information on the effect of the mutation on protein expression. A western blot study of WASp expression is possible but is restricted to certain research laboratories.
Certain diagnosis: genetics
The formal diagnosis of WAS/XLT is made by a classic sequencing study to identify pathogenic variants in the 12 exons/introns of the WAS gene. The detection of transmitting women will be an important element in family follow-up with family genetic screening and the proposal of prenatal counselling.
Differential diagnosis
The differential diagnoses of WAS/XLT syndrome are autoimmune or alloimmune thrombocytopenia and other constitutional thrombocytopenia.
Chronic immunological thrombocytopenic purpura (ITP) in boys is a diagnosis of exclusion, and Wiskott-Aldrich syndrome should be considered when thrombocytopenia occurs in boys under the age of one. ITP/XLT forms can be quite similar at diagnosis. The platelet volume of patients with WAS/XLT shows a small mean platelet volume (< 8 fL), which is almost pathognomonic for this syndrome, but is sometimes difficult to determine when thrombocytopenia is profound. A blood smear may then help to correct the diagnosis.
Platelet alloimmunisation should be considered in cases of thrombocytopenia occurring before the age of three months. It can sometimes be severe and lead to intracranial haemorrhages which, if they occur during the prenatal period, are likely to cause ventriculomegaly. The diagnosis is made by determining the platelet antigens of the child and their parents (HPA typing), by detecting anti-platelet antibodies in the maternal serum and by a cross-match test on the paternal platelets. A blood count and maternal autoimmune work-up should also be performed to avoid overlooking thrombocytopenia acquired through passive transfer of maternal autoantibodies secondary to maternal autoimmune disease. In both cases, thrombocytopenia will resolve after elimination of maternal antibodies at around six to nine months of age.
Other causes of hereditary syndromic thrombocytopenia should also be considered if there is associated extrahaematological involvement, including thrombocytopenia-absent radius syndrome (TAR syndrome), amegakaryocytosis with radio-ulnar synostosis, thrombocytopenia with oculo-oto-radial syndrome, Paris-Trousseau thrombocytopenia, and thrombocytopenia with velocardiofacial syndrome.
In this context, platelet volume will allow rapid differentiation of WAS/XLT from other conditions. Indeed, WAS/XLT is the only thrombocytopenia associated with a small platelet volume with ARPC1B deficiency. Other thrombocytopenias can be classified according to their platelet volume:
- ––Normal platelet volume: congenital amegakaryocytosis, familial autosomal dominant thrombocytopenia linked to chromosome 10, familial thrombocytopenia and predisposition to acute leukaemia related to mutation of AML1 (acute myeloid leukaemia), ANKRD26 (ankyrin repeat domain 26) thrombocytopenia, and thrombocytopenia with mutation in cytochrome C.
- ––Macro platelets: Bernard-Soulier syndrome, grey platelet syndrome, platelet pseudo-Willebrand, X-linked thrombocytopenia and GATA1, Mediterranean thrombocytopenia, MYH9 (myosin heavy chain 9) syndrome.
Finally, a WAS-like phenotype (thrombocytopenia with microplatelets) was recently described in a patient in whom a loss-of-function mutation in the WIPF1 gene was identified. This gene encodes the WIPF1 (WAS/WASL interacting protein family member 1) protein, which belongs to a family of WASp/WASL interacting proteins.
Treatments
Treatment must be carried out in conjunction with a unit specialising in immunology or haematology, due to the specific nature of their treatment.
Haemorrhages
Treatments for thrombocytopenia have improved over time, but some therapies should be avoided because of the deleterious complications of treatments usually used for immunological thrombocytopenia. Certain preventive measures against bleeding risks should be recommended, such as preventing trauma by avoiding certain high-risk sports (rugby, boxing, diving in a swimming pool, etc.) and wearing helmets for certain sports activities.
Polyvalent immunoglobulins at a dose of 0.8-1 g/kg, used in immunological thrombocytopenic purpura, are generally of little benefit. Splenectomy has historically been performed in these patients, as it is an effective way to increase the number of platelets. However, it is now clearly established that this treatment should no longer be used as a first line treatment due to the risk of infection, which can be fatal, in these patients [20]. If this treatment is envisaged or has already been carried out at the time of management, lifelong antibiotic prophylaxis with penicillin should be maintained with regular updating of the vaccination schedule. This procedure should also be avoided prior to HSC allografting, as it significantly increases the risk of post-transplant sepsis [24]. In the case of a patient with refractory thrombocytopenia, there is an indication for allograft transplantation, which is of major benefit compared to splenectomy. Severe refractory thrombocytopenia is currently classified as score 5, as it is most likely of autoimmune origin. Platelet transfusions should be avoided, except in the case of life-threatening emergencies, due to the risk of pre-transplant alloimmunisation, and the use of thrombopoietin receptor agonists should be favoured, while limiting the use of corticosteroids and immunoglobulins [26, 27]. Transfusion of platelets is of course possible but given the risk of developing anti-HLA antibodies in the context of underlying autoimmunity, these transfusions require prior treatment with corticosteroids to make them more effective and less deleterious. Platelet concentrates should be routinely irradiated (or, equivalently, treated with amotosalen and ultraviolet A) because of the constitutional cellular immune deficiency. As mentioned above, thrombopoietin receptor agonist therapy is appropriate as a transitional treatment prior to scheduled surgery (such as dental work) or pre-bone marrow transplantation. If the thrombocytopenia is major and resistant to the usual therapies, suggesting an autoimmune origin to the thrombocytopenia, treatment with immunosuppressants or anti-CD20 therapy should be discussed.
Infections
All patients with WAS should be treated for bacterial infections with a combination of antibiotic prophylaxis (cotrimoxazole [Bactrim®]) ± polyvalent immunoglobulin replacement (intravenous [IV] preferred). The decision to start antibiotic prophylaxis and/or introduce therapeutic antiviral and/or antifungal drugs depends on the frequency, severity and type of infections encountered, but does not depend on immune abnormalities which may not be marked on a brief immunological work-up (normal IgG level). These considerations apply to polyvalent immunoglobulin substitution. The IV route is preferred for classic forms of WAS, as thrombocytopenia is not compatible with subcutaneous administration. This is combined with the prevention of bacterial infections, mainly with encapsulated germs, by the addition of antibiotic prophylaxis based on daily cotrimoxazole, given the risk of systemic pneumococcal infections. When taken three times a week, this treatment also covers opportunistic infections such as pneumocystis and toxoplasmosis. It is recommended that the patient be vaccinated with inert vaccines and that the family be well-vaccinated with most of the recommended vaccines (pneumococcal, meningococcal, pentavalent, measles-mumps-rubella [MMR], influenza) [28].
Autoimmunity and inflammation
Treatments used in cases of autoimmunity or inflammation are essentially based on the use of immunosuppressants, primarily corticosteroids, but also anti-CD20 or other immunosuppressants, the prescription of which must be discussed with a specialised team, given the underlying immune deficiency. Anti-IL-1 drugs have recently been used in some complications of an auto-inflammatory nature. Autoimmune complications require immunosuppressive therapy, which can be challenging to use, since it is aimed at patients who already have a combined immune deficiency.
Autoimmune cytopenias may benefit from high-dose corticosteroid treatment for very short periods, often in combination with other anti-CD20 immunosuppressants and combined with IV immunoglobulin at 1 g/kg. Thrombopoietin receptor agonists may be combined if thrombocytopenia is severe (see above, bleeding). For autoimmune arthritis, treatment with immunoglobulins at 1 g/kg for two days with or without immunosuppressive agents (corticoids and/or cyclosporine) is usual. Cerebral vasculitis requires urgent immunosuppressive therapy.
Inflammatory bowel disease can be severe and should be treated like Crohn's disease with anti-tumour necrosis factor a (anti-TNFα).
All immunosuppressive treatments (steroids, immunosuppressive agents) should be administered with care by a specialist medical team. When introducing this type of treatment, the indication for medium-term HSC transplantation should be considered.
Severe eczema
This requires short-term topical or systemic treatment with steroids. The management of eczema is therefore based on the use of dermocorticoids, only on an ad hoc basis, giving preference to those with the lowest immunosuppressive effect and the greatest efficacy for the patient. Dupilumab can be used in case of failure of topical treatments and/or for corticosteroid sparing.
Role of haematopoietic stem cell transplantation
Historical data indicate that in the absence of curative treatment, patients with WAS have a short life span (15 years) [20], but the routine use of antimicrobial prophylaxis, prompt treatment of severe infections and autoimmune manifestations, and regular administration of immunoglobulins have led to a marked improvement in survival, thus most patients with WAS now reach adulthood.
However, the life expectancy and poor quality of life of patients with WAS, along with an increased risk of infections, autoimmune complications, severe bleeding episodes and malignancies, justify marrow transplantation, even for less severe forms such as XLT, as they are also subject to the same complications as those of patients with WAS [25]. Allogeneic HSC transplantation treatments for IDDs, including Wiskott-Aldrich, have improved markedly over the past 20 years, with >90% survival for paediatric patients and >85% for adults with PID [29]. The formal indication for patients with WAS depends on the severity of the score, but without a suitable donor, the prognosis is less favourable and life expectancy is reduced, especially in the case of malignancies. The first successful allogeneic HSC transplant for a patient with WAS took place in 1968, and several large series have been published since. For young children without a donor with a satisfactory HLA match, HSC transplantation with a haplo-identical donor gives satisfactory results (> 85%). If there is a clear indication, it should be performed as soon as possible with an HLA donor of the best possible match [10, 23]. Moratto et al. reported on a large retrospective multicentre study of 194 patients with WAS who received HSC transplantation between 1980 and 2009 [23]. The 194 patients received a total of 204 transplants (10 patients with a second transplant due to graft failure). The vast majority of patients (88.1%) received myeloablative conditioning with busulfan-cyclophos-phamide or busulfan-fludarabine. Survival at five years is 84%–but 89.1% for those transplanted after 2000. Transplants from a haploidentical intrafamilial donor or a placental blood unit were associated in this study with poorer survival. Nevertheless, the survival of transplanted children is currently improving, even for unrelated donor transplants in children over the age of five. Patients with a myeloid chimerism of <50% had a higher risk of persistent thrombocytopenia.
Since 2005, the international recommendations of the Inborn Errors Working Party of the European Society for Blood and Marrow Transplantation (EBMT) have recommended reducing the toxicity of myeloablative packaging by replacing high-dose endoxan with fludarabine. In addition, in the last ten years, some teams have replaced busulfan with its analogue, treosulfan, with a significant decrease in toxicity [31]. Thus, recent publications report very encouraging results in the transplantation of patients with WAS, both with pheno-identical donors and with placental blood units or haplo-identical related donors [32-34].
An HSC allograft is therefore the validated curative treatment for WAS. It is indicated in cases of severe forms (score 4 and 5) (figure 2); infectious complications which are poorly controlled despite optimal management, but especially autoimmune, inflammatory and/or neoplastic complications. It is indicated for patients with XLT or moderate WAS, if an HLA-compatible intrafamilial or unrelated donor is available [29, 30]. Moratto et al., in a recent publication, discussed the improved long-term quality of life of these patients after bone marrow transplantation, although a number of patients experience persistent autoimmunity/autoinflammation, depending on partial chimerism. The conditioning of the transplant is therefore essential in these patients in order to preserve their quality of life over the long term [29, 30].
Gene therapy
Gene therapy is currently still an experimental treatment, representing a possible alternative for patients who do not have compatible donors. It is performed in a very small number of specialised centres around the world, including the Necker-Enfants Malades hospital in Paris [35]. The first series was reported in 2013 by Aiuti et al. [36]. A lentiviral vector was used, along with reduced intensity conditioning prior to injection of the gene therapy product, in three patients. All showed improvement in clinical score, thrombocytopenia and a good level of immune cell transduction, with a good safety profile, especially in terms of genomic insertion of the transgene. A more recent trial with a lentiviral vector encoding human WAS cDNA, preceded by rituximab and busulfan and fludarabine conditioning, was conducted on eight patients with 100% survival [37]. The first results with a few years of follow-up showed a normalisation of T-cell function in vitro and the discontinuation of immunoglobulin supplementation in seven patients, as well as a reduction in infections. Prior to gene therapy, the platelet count was 20 G/L in all patients and > 100 G/L in two. No adverse reactions to the experimental drug or abnormal clonal proliferation or leukaemia were reported after gene therapy. Overall, the results of gene therapy show a satisfactory, if not perfect, correction of the immune deficiency and autoimmunity. It remains insufficient for the correction of thrombocytopenia, even if there is an improvement [36].
Perspectives and conclusions
All patients with WAS should receive specialised immuno-haematological follow-up through the Ceredih (www.ceredih.fr). Any chronic thrombocytopenia in boys should raise the possibility of a diagnosis of WAS/XLT, especially if it occurs early or is accompanied by a low MPV, CD8 lymphopenia, increased NK cells, hypereosinophilia or increased IgE. Many therapies can be considered in terms of prophylaxis, whether infectious (cotrimoxazole, polyvalent human immunoglobulin) or bleeding (thrombopoietin receptor agonists). In curative terms, HSC allograft marrow transplantation now offers an excellent prospect for cure.
Conflicts of interest
The authors declare no conflict of interest in relation to this article.