Hématologie
MENUVEXAS syndrome Volume 28, issue 6, Novembre-Décembre 2022

- DOI : 10.1684/hma.2022.1770
- Page(s) : 291-9
- Published in: 2022
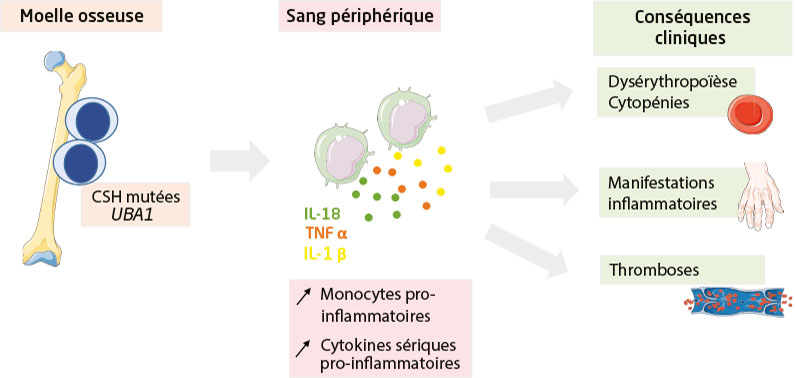
Le syndrome VEXAS (pour vacuoles dans les progéniteurs myéloïdes, enzyme activant l’ubiquitine E1, lié à l’X, manifestations auto-inflammatoires et somatiques) est un syndrome auto-inflammatoire récemment décrit, lié à des mutations somatiques de UBA1 (pour ubiquitin-like modifier activating enzyme 1). Ses principales caractéristiques cliniques sont hétérogènes, incluant une fièvre récurrente, des chondrites récidivantes, des lésions cutanées telles qu’une dermatose neutrophilique, une atteinte pulmonaire et oculaire, des thromboses veineuses, des atteintes ganglionnaires et des arthralgies. Le syndrome VEXAS est associé à une maladie hématologique dans 50 % des cas, le plus souvent un syndrome myélodysplasique ou une gammapathie monoclonale. Le traitement dépend de l’association ou non à une pathologie hématologique. Lorsque le syndrome VEXAS est associé à un syndrome myélodysplasique, les traitements hématologiques, en particulier l’azacitidine, semblent être parmi les plus efficaces, tandis qu’en l’absence de maladie hématologique, les inhibiteurs de kinase Janus semblent être les plus efficaces.