Hématologie
MENUPersonalised treatment for haemophilia Ahead of print

Haemophilia is a bleeding disorder resulting from an innate deficiency in factor VIII (haemophilia A) or factor IX (haemophilia B). Although there is currently no cure, the management of haemophilia has changed dramatically over the past decade with the development of improved treatments and new therapies. Since the 1960s, when cryoprecipitates containing factor VIII (about 3 to 5 IU/mL) were developed, the principle of haemophilia treatment has remained the same to this day; i.e. substitution of the missing clotting factor in one of two ways, either “on demand” when bleeding occurs, or “prophylactically” to prevent bleeding.
Prophylaxis: the gold standard in haemophilia treatment
Official bodies, including the World Health Organization (WHO) and the World Federation of Hemophilia (WFH), recommend prophylaxis as the optimal treatment strategy for severe haemophilia. The two main objectives of prophylaxis are to prevent serious, life-threatening bleeding, such as intracranial haemorrhages, and to prevent the development of haemophilic arthropathy, a major functional disability for haemophiliacs.
Several prophylactic protocols have been developed, with different perspectives and objectives. The first of these was initiated in Sweden in 1958. According to the Malmö protocol, prophylaxis aims at providing the patient with the “most normal life possible”by retaining the level of factor VIII (FVIII)/FIX above 1 IU/dL [1, 2]. This is a cumbersome and restrictive protocol, requiring several weekly intravenous injections of FVIII/FIX, and the prophylaxis should be started as early as possible, between the ages of one and two years old, often before any haemorrhagic event has occurred. Despite good clinical results and proven efficacy, this protocol has not been applied in other countries due to the high cost of injections and problems of availability of FVIII/IX concentrates in some countries [3]. It has also been met with reluctance from patients and their families due to the need for regular injections in children with haemophilia, the majority of whom have never suffered a bleeding episode.
These factors, which have prevented widespread use of the Swedish protocol, have led to the development of other prophylactic protocols, more appropriate for the individual needs of patients. The Dutch school thus advocated a scheme in which prophylaxis is initiated after the first haemarthrosis has occurred, through one or two weekly injections, with subsequent adjustments according to the patient's haemorrhagic tendencies, which may gradually intensify [4]. This was the first approach to personalised treatment for haemophilia.
Canadian teams then developed a “personalised” prophylactic protocol based on clinical criteria, in which the regimen is adjusted through progressive intensification of doses according to the patient's individual needs, based on bleeding observed under prophylactic treatment. The results of this strategy showed that only 16% of patients required a heavy regimen involving several injections per week, while 40% of patients received only one injection per week, significantly limiting the use of implanted ports. However, the effectiveness of this protocol has been questioned, as 40% of patients developed a target joint (i.e., at least three spontaneous haemarthroses within a period of six consecutive months) during the first five years of follow-up.
Thus, several questions currently remain concerning the optimal prophylactic regime:
- –what dose?
- –how often?
- –how should the characteristics of each patient, such as personal bleeding, joint condition, lifestyle, intensity of physical activity, degree of compliance with treatment, venous status, and response be considered as part of a strategy.
These parameters underline the relevance of an individualised, “tailor-made” prophylactic approach, which must be adapted to the needs of each individual and guided by objective, quantifiable and precise criteria, beyond intensification or reduction of a regimen based solely on the occurrence of symptomatic bleeding. Moreover, tailoring the prophylactic regimen based on symptomatic bleeding alone is not optimal to protect the joints.
One tool that can help define patients’ individual responses to different factor VIII or IX concentrates is a pharmacokinetic (PK) study, as significant variation in plasma levels of FVIII/FIX has been observed following injection of similar doses [5, 6].
Pharmacokinetic studies: one of the tools for tailored treatment
There are considerable differences in the pharmacokinetic profiles of patients who receive similar doses of FVIII or FIX, and variability in the half-life of FVIII has thus been reported by several authors. Following the injection of 30 IU/kg of FVIII in a child aged 0–6 years, the time required to obtain a residual rate of <1% is approximately 44 hours if the half-life is within the 5th percentile, compared to 78.1 hours in the 95th percentile, in other words, a difference of 34.1 hours. This difference is even greater for adolescents and adults (46.4 hours and 103.5 hours, respectively, a difference of 57.1 hours) [7]. Thus, individual variability regarding the half-life of factors could have a major impact on the prophylactic regimen.
It has also been shown that time spent below the critical threshold of 1 IU/dL exposes patients to a greater risk of bleeding. The length of time circulating FVIII/FIX activity remains above 1 IU/dL is primarily related to the half-life of the treatment, the frequency of injections, and patient compliance rather than the dose injected or the volume of distribution. Several parameters, including age and weight, contribute to this individual variability. Thus, children require higher doses of FVIII than adults because the clearance of FVIII decreases with age and thus its half-life progressively increases (this change in elimination with age is not observed for FIX) [8]. It may also be interesting to consider adjustments relating to weight with regard to obese patients.
While pharmacokinetic studies are a valuable tool, they do not take into account all the variables that influence the risk of bleeding. Joint condition is another major parameter that must be taken into consideration. One study tested two prophylactic regimens: an intense regimen with daily injections, and another with more widely spaced injections. The results show that, despite higher residual FVIII levels in the intense prophylactic group, the number of spontaneous haemarthroses was comparable in both groups, with a significant number of spontaneous bleeds in patients with target joints [9]. Similar results were reported in a gene therapy study, confirming the importance of including joint condition among the criteria for personalisation [10]. In this study, among patients with severe haemophilia B treated with a single dose of AAV-FIX vector, all patients reached plasma FIX levels of between 2 and 5 IU/dL. Prophylaxis may have been discontinued in some patients who no longer had spontaneous bleeding, while others, with target joints, were forced to continue treatment despite FIX levels ≥ 2 IU/dL, demonstrating the need for higher residual levels in subjects with target joints for effective prophylaxis.
These data strongly suggest that there is no “universal” residual threshold and that prophylaxis must be individualised, with individual thresholds determined according to the clinical and environmental characteristics of each patient and the properties of the factor concentrate being used.
The GENA-21 study was one of the first personalised prophylactic trials using PK. It was a multicentre, prospective, Phase IIIB study in patients with severe adult haemophilia A. The objective of GENA-21 was to test the efficacy of personalised prophylaxis based on PK data, using recombinant factor VIII (rFVIII) produced by human cells (h-Cl) [11]. Each patient received a PK evaluation with 60 ± 5 IU/kg rFVIII. Following this evaluation, patients entered the first phase of the study involving a standard prophylactic period with 30–40 IU/kg of the same FVIII, injected every other day – or three times a week – over a period of between one and three months. During this phase, patient PK data were analysed and a personalised prophylactic regimen, allowing for a residual rate of 1%, was determined for each subject. During the second phase of the study, patients were treated according to this personalised regimen for six months. The results of this study showed a significant reduction in the number of bleeding events during the personalised prophylactic phase, during which the injections were also spaced further apart in 58% of patients, and the amount of FVIII consumed was reduced by 8%. Seventy-three percent of the patients had no bleeding events. The PK study appears to be a promising tool that moves us closer to “precision medicine for haemophilia” by allowing us to improve prophylactic regimens for haemophiliacs based on their individual responses to each product. The target residual rate > 1%, which was shown to be effective in the Swedish clinical experiment and has been used as a basis in all PK studies, is nevertheless arbitrary.
Pharmacokinetic studies and new FVIII/FIX products with extended half-life
The importance of a personalised approach to prophylaxis became even more evident when FVIII/FIX concentrates with a prolonged half-life were introduced into the therapeutic arsenal. An extended half-life not only allows for longer injection intervals, but may offer better anti-haemorrhagic protection by retaining similar injection intervals and residual rate targets better suited to the needs of each individual. Such an approach could significantly reduce the number of haemorrhagic accidents, if the economic conditions for treatment allow.
The results of the Phase III PROLONG-9FP studies (CSL654-3001 and CSL654-3002) were the first to validate this approach [12, 13]. These studies tested the efficacy and safety of FIX-albumin (rFIX-FP) prophylaxis using different regimens involving injections every 7–10 or 14 days in patients aged 12 to 61 years with plasma FIX levels < 2%. The average half-life of rFIX-FP was 102 hours, which is 4.3-fold longer than that of conventional FIX. All patients included in the study had a PK assessment. The mean residual rates observed were 20% and 12% for the 40 IU/kg/7 day and 75 IU/kg/14 day groups, respectively The number of annual spontaneous bleeds was zero for all groups (injections every 7–10 and 14 days). Some patients with a reduced bleeding profile were able to receive prophylaxis every 21 days without spontaneous bleeding events.
Following these observations, several authors developed Bayesian PK evaluation programmes, adapted to the different FVIII and FIX products, in order to support the personalisation of anti-haemophilic treatments. These tools, which require only two or three blood samples (rather than the 10 or 12 samples based on previous PK, which is very difficult to perform clinically), make it possible to perform prophylactic regimen simulations with various doses and injection intervals in order to achieve predefined residual target rates [14]. Maintaining a higher residual level in young patients with very active lifestyles using treatments with a prolonged half-life could be of particular interest, to better protect them against the risk of bleeding while allowing them to maintain their activities. In order to optimise the performance of PK tools, a recent study defined minimum residual values in different clinical contexts using the Delphi method with the participation of 15 international experts. The experts defined three residual levels corresponding to different patient profiles, activities and bleeding phenotypes [15]:
- –1–3% for patients with an attenuated haemorrhagic phenotype receiving prophylaxis,
- –3–5% for active patients and for those with moderate arthropathies,
- –5–15% for patients involved in at-risk physical activities or with target joints and severe arthropathies.
In an interventional study, the same authors then used these three residual levels to individually tailor patient prophylaxis using a PK tool [16]. During the study period, they considered a residual rate < 3% to be at risk of bleeding, a rate of 3–15% as appropriate for moderate physical activity and values >15% as “without danger” regardless of the physical activity in question. This study reported no bleeding events during the personalised prophylactic phase. This type of approach, using high target residual values (> 15%), is certainly very safe for the joints, but increases the cost of prophylaxis, taking us significantly further away from the initial cost estimates published by Carlsson et al.[17] showing a 20–30% reduction in the quantity of FVIII/FIX concentrates used under personalised prophylaxis. The authors suggested that PK-guided prophylaxis represents a true cost benefit, in addition to more effective coverage for patients. Based on these observations, “residual clotting capacity”, rather than residual FVIII, is probably an individual concept that should be measured.
Can we move towards more effective tailored treatment?
A new model combining PK studies with pharmacodynamics (PD) has been developed in an attempt to better tailor the prophylactic regimen to the individual needs of each patient [18]. PK studies address what happens to a treatment product in the body, while PD addresses the effect it has on the body (in the case of haemophilia, improved clotting ability). The authors proposed, using the thrombin generation test, to assess pharmacodynamics in combination with PK adapted to the FVIII concentrate used. The thrombin generation test is a comprehensive haemostasis test that takes into account not only the pro-coagulant effect of FVIII or FIX but also the impact of all other pro-coagulant proteins and natural coagulation regulators such as antithrombin, the protein C system and tissue factor pathway inhibitor (TFPI). By developing a combined PK + PD tool, the authors improved the accuracy of the simulations, compared to those based on the PK tool alone. This interesting and innovative approach needs to be tested prospectively in new clinical studies. Its effectiveness and efficiency should be compared to personalised prophylaxis using PK analysis alone.
Is there a place for tailored medicine using new non-substitutive therapeutic approaches?
Emicizumab (Hemlibra®) was the first non-substitutive treatment available on the market. It is a humanised bispecific monoclonal antibody that mimics the clotting function of activated FVIII (FVIIIa) by binding to Factor IXa and Factor X. Emicizumab does not show any structural similarity or share any sequence homology with Factor VIII, and is therefore not neutralised by inhibitors directed against Factor VIII. It is administered by weekly subcutaneous injections and is indicated for prophylaxis of bleeding episodes in patients with haemophilia A with or without anti-factor VIII alloantibodies known as inhibitors [19].
The administration of FVIII concentrates may induce the development of anti-FVIII alloantibodies in some patients, particularly in patients with severe haemophilia A. The development of an anti-FVIII inhibitor is the biggest challenge in the treatment of haemophilia, as inhibitors compromise the effectiveness of replacement therapy and significantly increase functional disability related to haemophilic arthropathy and mortality. The therapeutic management of haemophilia A with inhibitors usually consists of two components:
- –an Immune Tolerance Induction protocol (ITI) using injections of FVIII concentrates aimed at eradicating the inhibitor,
- –administration of so-called “bypass agents” to treat and/or prevent bleeding.
The two by-pass products available on the market are recombinant activated factor VII (rFVIIa, Novoseven®) and activated prothrombin complex concentrate (aPCC, Feiba®). These two products have different, multiple and complex mechanisms of action, but they are both effective as they ultimately support the generation of thrombin in patients. In a surgical context, they both exhibit 85% efficiency. However, no routine test has been approved to monitor their effectiveness and adapt their dosage to patient needs. Patients with severe haemophilia often have to undergo orthopaedic surgery at an advanced stage of their arthropathy. To ensure effective haemostasis during these major surgical procedures, it is important to know the individual patient's response and to objectively evaluate the efficacy of both products prior to surgery. Thrombin generation testing has been shown to be an interesting biomarker that may be used to assess a specific patient response to by-passing agents and thus tailored treatment in the context of planned surgery [20]. Similar results have also been reported with thromboelastometry [21]. Global haemostasis tests, such as thrombin generation testing and thromboelastometry, are therefore biological tools that can be used for the personalised prescription of bypass therapy in haemophiliacs with inhibitors.
Emicizumab was recently introduced to the list of treatments for haemophilia A alongside inhibitors, and is indicated for prophylaxis of bleeding episodes.
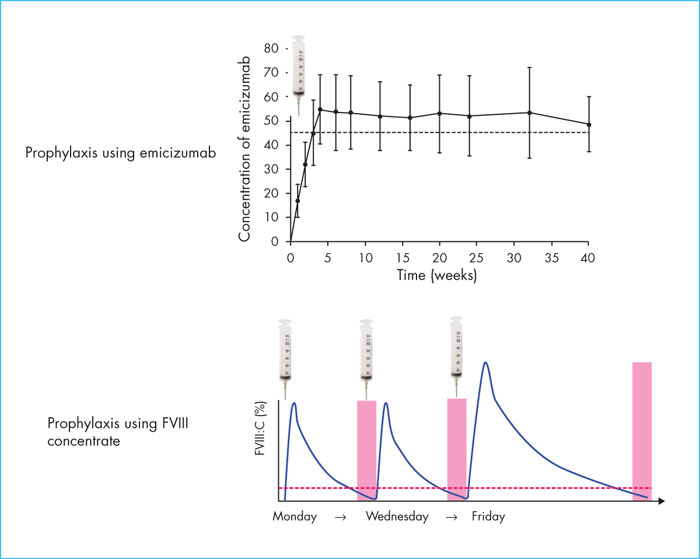
Prophylaxis with emicizumab results in stable and prolonged clotting, which is fundamentally different from conventional prophylaxis with FVIII concentrate or bypass agents that result in a sawtooth haemostatic profile (Figure 1). The level of clotting achieved with emicizumab has not been fully normalised but is usually higher than that provided by the residual FVIII level. In the case of intercurrent bleeding, treatment is provided through the administration of FVIII or bypass agents. Serious thromboembolic events have been reported in patients with inhibitors during concomitant use of emicizumab and aPCC [22]. These events were observed when aPCC was prescribed at a mean dose >100 units/kg/day for more than one day, i.e., the usual dose of 75 units/kg every 8–12 hours. There is also a possibility of hypercoagulability when rFVIIa or FVIII is administered with emicizumab, based on pre-clinical studies. Emicizumab increases clotting capacity; therefore, the dose of clotting factor required to achieve haemostasis may be less than that used in the absence of prophylaxis with emicizumab. Customised prescriptions are essential in this context of concomitant use of several procoagulant products, in order to ensure effective haemostasis without inducing hypercoagulability that may cause thrombotic complications. A published clinical case study illustrates this personalised approach [23]. This relates to a patient with severe haemophilia A who was receiving prophylactic emicizumab and who presented with a retroperitoneal haematoma upon spontaneous rupture of a malformed artery at the upper pole of the left kidney. The patient received standard treatment with aPCC due to an insufficient individual response to rFVIIa. The combined effect of emicizumab and aPCC was evaluated in vitro prior to the event, with increasing doses of aPCC. Doses of aPCC between 15 and 25 U/kg normalised thrombin generation in vitro without inducing hypercoagulability. The patient therefore received treatment with aPCC at 25 U/kg, followed by twice-daily doses of 15 U/kg for three weeks. Bleeding was controlled with medical treatment and no thrombotic complications were reported in connection with this treatment. This case illustrates the fact that the personalised approach using controlled laboratory techniques makes it possible to better secure the use of new non-substitutive treatments.
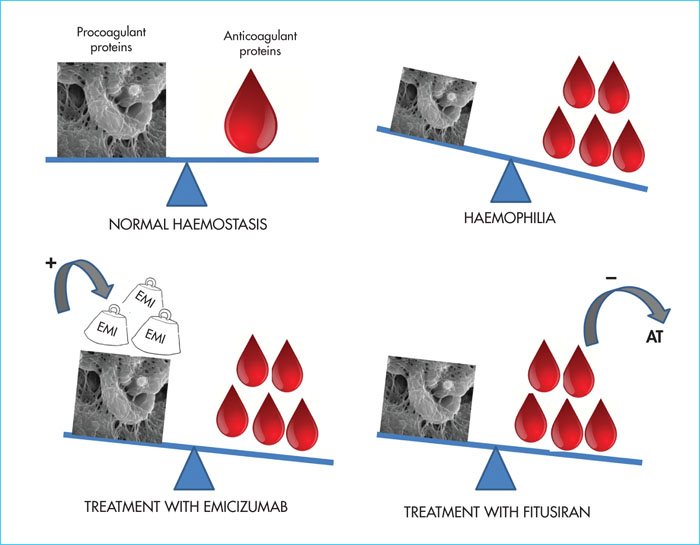
Another non-substitutive treatment under development is fitusiran. This is an interfering RNA, administered subcutaneously with a half-life of 30 days, for the treatment of haemophilia and other rare bleeding disorders. This molecule aims to correct coagulation defects by altering the balance between procoagulant coagulation factors and inhibitors that are natural anticoagulants(Figure 2). It reduces the expression of antithrombin, the most potent endogenous anticoagulant, and creates an acquired, controlled and transient antithrombin deficiency [24]. Two doses of fitusiran, 50 and 80 mg/month, have been tested in Phase II studies; in both cases, an 80% reduction in antithrombin was observed and 48% of the patients had no bleeding events [25]. The Phase I and II studies clearly demonstrated that a decrease in circulating antithrombin was consistently accompanied by an increase in thrombin generation. In most cases, a fixed monthly dose was effective during the Phase II studies. Nevertheless, a fatal cerebral venous thrombosis occurred in one patient who received fitusiran and FVIII concentrate (31-46 U/kg) following intercurrent bleeding. Recommendations, based on the in vitro evaluation of thrombin generation, were then published for the prescription of factor concentrates in combination with fitusiran [26]. It is recommended that antifibrinolytics be avoided in combination with fitusiran. In case of a haemorrhagic event in a patient receiving prophylactic fitusiran, low doses of concentrates are preferred: FVIII at 10-20 IU/kg, FIX at 20-30 IU/kg, aPCC 3 at 0-50 U/kg and rFVIIa ≤ 45 μg/kg [26].
Marstacimab (PF 6741086) is a humanised anti-TFPI monoclonal antibody which, like fitusiran, exerts a procoagulant effect by inhibiting an endogenous anticoagulant, TFPI. It is the main inhibitor of the clotting initiation phase. As with emicizumab, the arrival of these two innovative products, currently under development for prophylaxis, on the market will raise issues on the management of intercurrent bleeding that occurs under prophylaxis. Their concomitant prescription with other procoagulant treatments will require customised doses to ensure their effectiveness and avoid thrombotic events if possible. It should be noted that development programmes for the other two anti-TFPI monoclonal antibodies were recently halted due to serious thrombotic events.
Personalised treatments because every patient is different
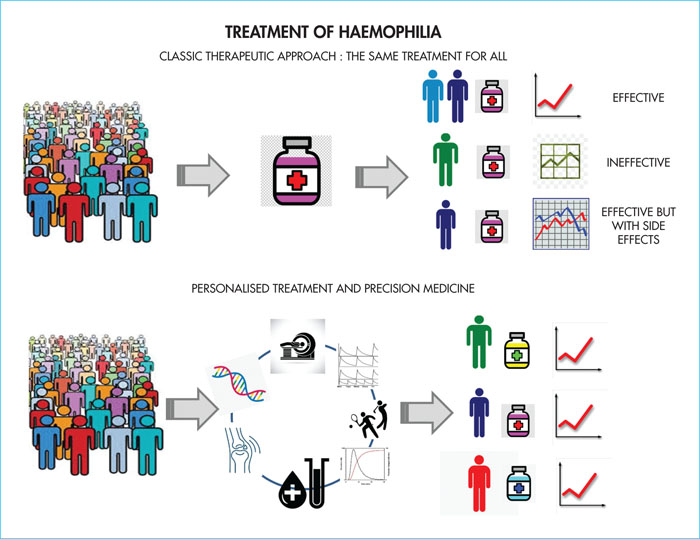
Despite similar plasma FVIII levels, 10–15% of patients with severe haemophilia A have a moderate/minor haemorrhagic phenotype, with rare bleeding episodes, reduced factor intake and no haemophilic arthropathy. The concomitant expression of constitutional thrombophilia, such as the Leiden mutation or involvement of hepatitis B or C viruses or HIV, are thrombotic risk factors that can alter the clinical expression of haemophilia. The genotype is another determinant of the haemorrhagic phenotype. It has been shown that certain mutations such as missense mutations, small deletions/insertions and splice site mutations are associated with minimal circulating levels of factor VIII, providing some protection against bleeding [27]. On the other hand, the levels of clotting factors and inhibitors are highly variable, and can vary from one subject to another by as much as three-fold, while remaining within normal values (as a reminder, normal levels of most factors are between 50 and 150%). Thus, FVIII or FIX activity is not the only determinant of an individual's overall coagulation capacity. All these considerations are changing our vision of haemophilia treatment and are moving towards the need for precision personalised medicine, integrating genomics and lifestyle. This will allow optimal dosages of different therapeutic molecules to be determined for each patient according to their individual response, securing concomitant use of several procoagulant factors, giving patients the benefit of customised doses adapted to their coagulation capacity(Figure 3).
The treatment of paediatric patients and adult patients with certain co-morbidities should be carefully monitored, evaluated and adapted. The principle of RNA interfering with antithrombin messenger RNA has been validated in adults with mature and normal livers. The administration of fitusiran to patients with a history of untreated hepatitis C led to an increase in liver enzymes and discontinuation of treatment in these patients during the Phase I study [25]. Newborns and infants with “immature” livers may be more vulnerable to this type of side effect. The efficacy of emicizumab has been validated in children under 12 years of age [28]. Nevertheless, only eight children between one and two years of age were included in the study and there are no data on infants < one year of age with immature livers, in whom the levels of FIX and FX are different from those found in adults. These differences may alter the PK and PD of emicizumab in this particular population and may require adjustments based on liver maturity.
Customisation may also concern the choice of molecules adapted to each patient. After 50 years of treating haemophiliacs with FVIII and FIX concentrates, we are at a turning point with the advent of improved recombinant molecules and non-substitutive therapies that are a source of hope for physicians and patients. These new approaches open up the prospect of reducing the occurrence of the main complication of haemophilia in 2020: the development of inhibitors. The “personalised” choice to use these approaches may be of interest and justified in patients with mutations and other constitutional and acquired features, at high risk of developing inhibitors.
Is a cure for haemophilia in sight?
Recent results from gene therapy clinical trials are very promising and offer hope for a cure for haemophilia A and B [29, 30]. 2020 is likely to be a turning point, with the possible arrival on the market of the first gene therapy product for haemophilia A. Valoctocogen roxaparvovec has resulted in stable FVIII expression in approximately 20% patients followed for three years, with a more than 95% decrease in the need for FVIII concentrates. The results obtained with regard to haemophilia B are also very promising showing stable plasma levels at 5–10% with wild type FIX and a possibility of normalisation of the FIX level at between 35–50% and even beyond when the transgene used is FIX-Padua, a variant with increased specific activity. Will personalisation still be relevant when gene therapy becomes part of the standard treatment for haemophilia? If we look closely at the results of the Phase III studies, the variability of results, the predictability of rates achieved depending on the amount of vectors administered, the durability at the individual level, and the risk of hepatotoxicity suggest that the personalised approach to haemophilia treatment may still play a role in the era of gene therapy.
Conflicts of interest
None of the authors have any conflicts of interest to disclose.