Hématologie
MENUPathophysiology and abnormalities of haemostasis in intensive care patients with COVID-19 Ahead of print

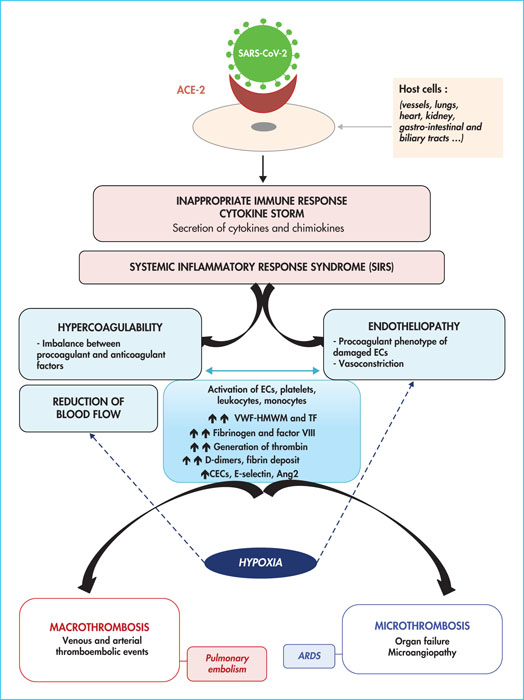
The respiratory infection caused by coronavirus 2 (SARS-CoV-2), now named COVID-19 (for coronavirus disease 2019), was first identified in Wuhan, in the Hubei province of China, in December 2019 and has since spread worldwide [1, 2]. Following an average incubation period of five days, some patients infected with SARS-CoV-2 develop an influenza-like syndrome with fever, dyspnoea and a cough [1, 2]. Extra-respiratory signs affecting the kidneys, brain and heart have also been observed. Other infected individuals, in contrast, are asymptomatic. In some patients, an inadequate immune response leads to an amplification of the inflammatory response and a worsening of the pneumopathy, 7–10 days after the onset of the first symptoms. Around 5–10% of COVID-19 patients develop severe pneumonia, including parenchymal and alveolar involvement leading to hypoxaemic acute respiratory failure, and sometimes severe acute respiratory syndrome (SARS) [2]. Patients with a severe form of COVID-19 are therefore likely to develop hypoxia (requiring invasive ventilation support), excessive inflammation, thrombotic complications such as pulmonary embolism (20–30% of cases), deep vein thrombosis (DVT), arterial thrombosis, and unusually frequent thrombi which can obstruct dialysis circuits and extracorporeal membrane oxygenation (ECMO) circuits [3-7]. In addition, microvascular thrombosis, acrosyndrome, and capillary leakage syndrome affecting the lungs, kidneys and heart, potentially complicated by multivisceral failure, have been described [4, 5]. In this review, we summarise the complex mechanisms that may explain the disorders of haemostasis observed in patients with severe forms of COVID-19 hospitalised in intensive care (figure 1)[8].
Inappropriate immune response and systemic inflammatory response syndrome
The receptor that allows SARS-CoV-2 to enter the host cell is angiotensin-converting enzyme 2 (ACE2), which is highly expressed in the arterial and venous vascular endothelium of many organs, including the pulmonary alveoli [9-11](figure 1). Damage to the lungs caused by SARS-CoV-2 induces injury to bronchial epithelial and endothelial cells, and causes alveolar infiltration by inflammatory cells, leading to an increase in circulating cytokines – interleukin 1β (IL-1 β), IL-2, IL-6, IL-7, IL-10, granulocyte colony-stimulating factor (G-CSF), interferon γ (IFNγ)-induced protein 10 (IP10), monocyte chemoattractant protein (MCP1), macrophage inflammatory protein type 1A (MIP1A), tumour necrosis factor α (TNFα) [12-14], and chemokines [15]. IL-6 plays an important role in the activation of endothelial cells in the early phase of inflammation, and a significant increase in IL-6 is reported in patients with a severe form of COVID-19, associated with a poor prognosis [12-14]. In its severe form, SARS-CoV-2 infection is associated with an inappropriate immune response, also known as a cytokine storm, marked by cytokine release syndrome and systemic inflammatory response syndrome (SIRS), an intense inflammation that causes endothelial dysfunction [4, 13, 14](figure 1). Furthermore, the production and activity of type I IFNs are significantly decreased in the severe form of COVID-19, leading to an ineffective and pathological inflammatory response [16].
Relationship between hypoxia, inflammatory response, hypercoagulability and endotheliopathy
Compared to other viral lung diseases and sepsis, the haemostatic disorders reported in patients with severe COVID-19 present specific features related to severe hypoxia, thrombo-inflammation, a state of hypercoagulability and endothelial dysfunction [4, 5, 9, 17](figure 1). The profound hypoxia observed in pulmonary capillaries in patients with COVID-19 increases vasoconstriction and reduces blood flow, contributing to endothelial dysfunction [5, 9, 10]. Hypoxia can promote a shift from the antithrombotic and anti-inflammatory phenotype of endothelial cells to a procoagulant and proinflammatory phenotype, mainly by altering transcriptional factors such as the early growth response gene 1 (Egr1) and hypoxia-inducible factor 1 (HIF-1), previously described for other ARDS [5, 18]. HIF1 synthesis modifies and activates the synthesis of tissue factor (TF) and plasminogen activator inhibitor 1 (PAI-1) [18]. In contrast, the conversion enzyme is responsible for the hydrolysis of angiotensin I to vasoconstrictor angiotensin II and degradation of the vasodilator, bradykinin. Interference of the conversion enzyme by SARS-CoV-2 results in a generalised vasospasm [17]. Secondly, pro-inflammatory cytokines involved in COVID-19 induce dysfunction of the endothelium, resulting in massive release of vonWillebrand factor high-molecular weight multimers (VWF-HMWM) that are hyperadhesive to platelets and the subendothelium, as well as overexpression of TF [5, 13, 14, 17, 19]. SARS-CoV-2 infection induces the recruitment of mononuclear cells to the alveolocapillary barrier. TF expression on the surface of activated mononuclear cells promotes thrombin generation, as well as platelet activation and interaction with the activated endothelium. The release of TF and neutrophil extracellular traps (NETs) into the extracellular medium allows coagulation to be initiated by the activated TF/factor VII axis. In addition, NETs contribute to the activation of FXII. Excessive concentrations of thrombin are subsequently generated [20](figure 1). This state of hypercoagulability is thus enhanced by an increase in procoagulant factors (factors V and VIII, fibrinogen), an overload of the coagulation regulatory systems (antithrombin, proteins C and S) [5, 17, 19, 21-24], the activation of platelets and, finally, their interaction with the activated endothelium. Although a lupus anticoagulant is found in 80% of patients [5, 17, 25, 26], its association with increased thrombotic risk has not been established [25]. Finally, activation of the endothelium, secondary to SARS-CoV-2 infection or induced by the membrane activation complex, promotes coagulation activation and the interaction between the endothelium and circulating platelets [17,27]. Factor VIII coagulant activity, von Willebrand factor antigen [5], and circulating endothelial cells (CECs) are increased in patients infected with SARS-CoV-2 [28]. The increase in CEC levels reflects endothelial dysfunction in severe forms of COVID-19 [29]. Moreover, the increase in E-selectin and angiopoietin 2 concentrations correlates with the severity of the infection, reinforcing the hypothesis of pulmonary vascular involvement in COVID-19 [30].
Systemic thrombosis of macro- and microcirculation
Thrombosis of the macrocirculation and microcirculation, and diffuse endothelial damage may explain the multisystem symptomatology of SARS-CoV-2 infection. Nearly 45% of patients with a severe form of COVID-19 develop thrombotic complications, despite anticoagulant treatment with at least thromboprophylactic dosage, administered upon admission to the hospital or medical intensive care unit [3, 5-7, 31-33](table 1 table 1). Altogether, slow blood flow (induced by vasoconstriction and venous stasis), endothelial aggression, hypercoagulability, and Virchow's triad protagonists, account for the very high risk of venous thrombosis in these patients [34, 35]. The occurrence of venous macrothrombosis (DVT and pulmonary embolism) is encouraged by excessive thrombin generation aggravated by an imbalance between procoagulant and anticoagulant factors, whereas arterial macrothrombosis (mainly strokes) can be related to an increase in VWF-HMWM and platelet activation [36](figure 1). Interestingly, systemic microthrombosis is reported in severe forms of COVID-19 and is sometimes complicated by multivisceral failure. In contrast to the coagulation disorders seen with sepsis, platelet and coagulation factor consumption is less frequent in patients with severe COVID-19, suggesting that disseminated intravascular coagulation (DIC) is not a common complication of SARS-CoV-2 infection [5-7, 19, 28, 37]. Pulmonary microthrombosis is the pathophysiological basis for ARDS associated with SARS-CoV-2 (figure 1).
Patients with severe forms of COVID-19 have altered alveolar and pulmonary microcirculation associated with platelet/VWF-HMWM interaction related to the injured endothelium. In addition, impaired fibrinolytic function can lead to abnormal accumulation of fibrin in the alveolar spaces with the formation of localised or disseminated microthrombi, readily promoted by vasoconstriction and the reduced blood flow induced by deep hypoxia in the pulmonary capillaries [4-6, 38]. However, some mechanisms associated with SARS-CoV-2 infection have not been elucidated, and it is difficult to predict the presence of severe coagulation abnormalities in patients with severe ARDS. Bleeding complications are rare in patients with severe COVID-19 (less than 3% of patients), with the exception of those related to the use of anticoagulants [5].
Laboratory monitoring of haemostasis and anticoagulation
The laboratory monitoring of haemostasis in patients with severe forms of COVID-19 relies mainly on the measurement of prothrombin time, fibrinogen, D-dimer levels and platelet count. Increased D-dimer levels have been identified as a predictive marker for the development of ARDS and the severity of the disease (admission to intensive care, prognosis) [2, 5, 14, 34, 35, 37, 39-42] (table 2 table 2). Elevated fibrinogen and D-dimer levels reflect the patient's inflammatory state as well as the state of hypercoagulability, from the very onset of the disease [43]. An increase in D-dimer levels (>3,000 ng/mL, six times above normal) and fibrinogen levels (>8 g/L) is associated with a higher risk of mortality and thromboembolic complications [44]. Fibrin monomers, specific markers of consumption syndrome, do not appear to be increased in COVID-19 [28, 30]. Other coagulation biomarkers increased levels, such as factor VIII and von Willebrand factor, are associated with disease severity and are predictive of increased oxygen requirements in patients [45]. Viscoelastic tests and quantification of the fibrinolytic function could be useful in defining a state of persistent hypercoagulability and could therefore predict thrombotic risk in these patients [20, 46, 47]. Despite standard thromboprophylaxis with low-molecular weight heparin (LMWH) or unfractionated heparin (UFH), the prevalence of thrombotic events has been shown to be unusually high, particularly in intensive care units. Therefore, thromboprophylaxis, preferably with LMWH rather than UFH in the absence of contraindications, should be systematic. An increased dosage, taking into account individual risk factors (body mass index, personal history, comorbidities, etc.), should be discussed [4-7, 19, 32, 35, 44, 48]. The benefit/risk ratio of curative doses of anticoagulant, taking into account certain biomarkers or clinical risk factors, has not yet been prospectively demonstrated, with the exception of patients with a confirmed or strongly suspected diagnosis of a thromboembolic event, or patients on ECMO. Furthermore, in patients treated with curative UFH, therapeutic goals in terms of anti-Xa activity may be difficult to achieve, considering major inflammatory syndrome, and require very high, weight-based doses [42, 49].
Conclusion
The links between the different concepts involved in the pathophysiology of SARS-CoV-2 infection in terms of immune response, inflammation, endothelial injury and hypercoagulability remain to be established. In practice, it has now been shown that patients with COVID-19 are at high risk of thrombosis. Routine haemostasis tests alone are not sufficient to predict the risk of macro- or microcirculatory thrombosis induced by SARS-CoV-2 infection. D-dimers could be considered as a biomarker of inflammation in order to better predict thrombotic risk and to assess the prognosis of individual patients. In the absence of a risk of bleeding, when thrombosis has been established, patients with a severe form of COVID-19 should receive enhanced prophylactic-dose anticoagulation while awaiting the results of randomised studies.
Conflicts of interest
None of the authors have any conflicts of interest to disclose.