Hématologie
MENUNext-generation sequencing in patients with hairy cell leukaemia (HCL) Ahead of print

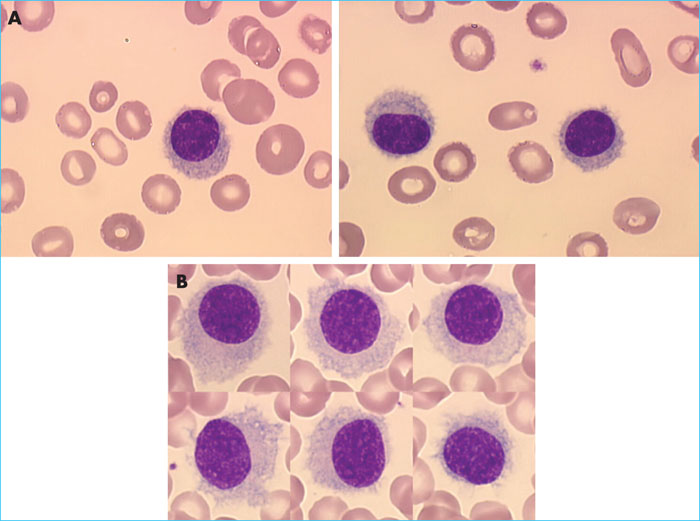
Hairy cell leukaemia (HCC) is a rare mature B-cell lymphoid hemopathy. It accounts for about 2% of all leukaemias and occurs preferentially in men over the age of 50. The variant form of the disease, known as HCL-v, constitutes – in contrast to HCL – a provisional entity in the 2016 World Health Organization classification [1] and represents around 10% of cases of HCL. It is necessary to differentiate between the two forms of the disease, given the different prognosis and treatments. Diagnosis of HCL is based on the identification in the blood and/or marrow of abnormal hairy lymphoid cells (Figure 1A). Flow cytometry (FCM) examination of the lymphoid cells in the blood and/or marrow reveals the presence of mature monotypic B lymphocytes which express at least three of the following four markers: CD103, CD123, CD25 and CD11c [2]. It is necessary to look for the BRAFV600Emutation, which was identified in 2011 in exon 15 of the BRAF gene [3]. There are undeniably rare forms of HCL that are negative for BRAFV600E, which have a poor prognosis and for which BRAF inhibitors are not authorised as treatment. HCL-v is differentiated from HCL by the presence of a prominent nucleolus (Figure 1B), a typical lack of expression of markers CD123 and CD25 and a lack of BRAF mutation. The first-line treatment for HCL is based on purine (PNA) analysis, and second-line treatment consists of a combination of PNA and rituximab. In the event of a later relapse, treatments are less well codified [4]; BRAF inhibitors, moxetumomab pasudotox, a monoclonal anti-CD22 antibody coupled with Pseudomonas toxin [4], and BTK inhibitors may be therapeutic options.
The genetics of hairy cell leukaemia
At the time BRAFV600E mutation was initially described, it was identified in all patients with HCL but not in patients with any of the other chronic B lymphoproliferative disorders. However, the BRAFV600E mutation is not specific to HCL; it is also found inmore than 50% of melanomas, Chester-Erdheim's disease, solid colorectal and lung tumours, albeit less frequently, as well as other haematological malignancies: chronic lymphocytic leukaemia (CLL) and multiple myeloma (MM) of the bones. Furthermore, the BRAFV600Emutation is not present in a subgroup of HCL patients with a poor prognosis, corresponding to patients with IGHV4-34 rearrangement [5, 6]. This rearrangement is absent in HCL-v, which is why the activating mutations of MAP2K1, which encodes the MEK protein, are nevertheless identified in more than a third of cases [7]. Alternative BRAF mutations have been identified in exon 11 [8]. BRAF and MAP2K1 mutations are mutually exclusive. The development of high-throughput sequencing techniques has increased our knowledge of the mutation profile of HCL and HCL-v [3, 7, 9-12] and has identified additional recurrent mutations associated with BRAFV600E. These mutations could play a role in the initiation and/or progression of the disease. All the genetic variants of interest, as well as their frequency, that have been reported in the various published high-throughput sequencing studies are presented in table 1.
MAP kinase pathway
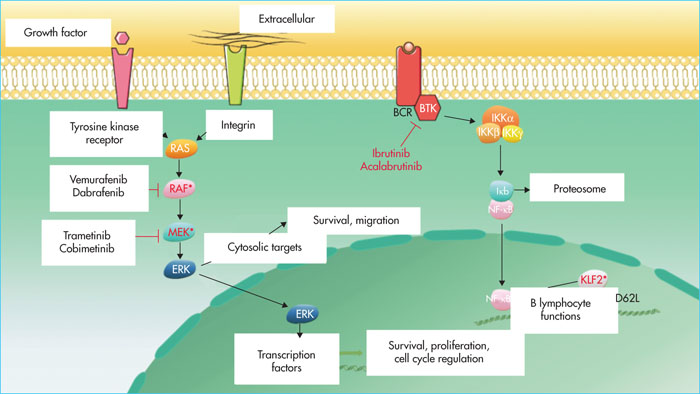
BRAF encodes for a serine-threonine kinase(B-Raf proto-oncogene, serine/threonine kinase) involved in the MAP (mitogen-activated protein) kinase (MAPK) pathway. It phosphorylates MEK (mitogen-activated extracellular signal-regulated protein kinase), which in turn phosphorylates ERK (extracellular signal-regulated kinase). These proteins, located in the nucleus, activate transcription factors that induce cell proliferation, survival, and invasion signals (Figure 2). The BRAFV600Emutation, located in the kinase domain, constitutively activates BRAF[3]. MAP2K1 mutations, located in the negative self-regulatory domain of the gene, also activate MEK1. The location of the mutation may increase sensitivity to MEK inhibitors or, on the contrary, induce resistance [7]. The MAPK pathway is also involved in the regulation of the expression of cyclin D1 and the p27 protein, which are involved in cell cycle control and are deregulated in HCL [13-16]. The MAPK pathway is involved in the hair morphology of hairy cells; the use of BRAF inhibitors, notably vemurafenib, can reverse the villous phenotype and can decrease the expression of ACTB (b-actin) and LST1, which code for proteins involved in cytoskeleton formation [17]. The identification of activating mutations in the MAPK pathway has logically led to the introduction of BRAF inhibitors (vemurafenib and dabrafenib) with or without MEK inhibitors (trametinib and cobimetinib) as treatment for HCL. An antileukaemic effect of these treatments, both in vitro and in vivo, on hairy cells carrying the BRAFV600E mutation has been demonstrated [17]. Patients may develop resistance to BRAF inhibitors, but the mechanisms of resistance remain poorly understood. In melanomas, they lead to a reactivation of the MAPK pathway or abnormal activation of the phosphatidylinositol kinase 3 (PI3K)/AKT pathway. The action of BRAF inhibitors can be circumvented by the emergence of an activating mutation of another pathway effector, such as NRAS, KRAS[12] or MAP2K1, by amplifying the number of copies of the mutated BRAF gene or by alternative splicing of BRAF, generating a protein that is insensitive to specific inhibitors [18]. In patients with de novo resistance to vemurafenib, the simultaneous presence of an IRS1 activating mutation associated with a deletion in NF1 and NF2 has been identified [12]. IRS1 may activate the MAPK and PI3K/AKT pathways by transmitting a signal through the insulin growth factor receptor (IGFR1). NF1 is a negative regulator of RAS proteins. In order to improve the response to BRAF inhibitors and to circumvent resistance mechanisms, strategies based on the simultaneous use of BRAF inhibitors and MEK1 (NCT02034110), or the combination of vemurafenib with a recombinant human anti-CD20 type II monoclonal antibody, obinutuzumab (NCT03410875), are currently being investigated.
Cell cycle
Several genes (CDKN1B, CCND1, CCND3 and TP53), encoding proteins involved in cell cycle regulation, are deregulated in HCL. Inactivating mutations of CDKN1B, again associated with the BRAFV600Emutation, are identified in 10–15% of HCL cases [9, 10, 12]. CDKN1B encodes for p27, which controls the progression of cells in the cycle by binding and inactivating cyclin-dependent kinase (CDK) complexes. p27 is known to be deregulated in many cancers, either due to a decrease in expression or a change in location: this deregulation is associated with poor prognosis. In most cancers, CDKN1B mutations are identified with a frequency of less than 5%; these are identified in 15% of HCL cases and neuroendocrine tumours of the intestine [9]. The loss of CDKN1B expression may be necessary for tumour development, allowing hairy cells to escape senescence that may be induced by BRAF mutations [19]. p27 also has an antagonistic activity towards cyclin D1 as it inhibits the activity of the cyclin D1-Cdk4 complex [20]. Cyclin D1 is over-expressed in HCL [21, 22] and enables the phosphorylation of retinoblastoma proteins, allowing the release of the transcription factor E2F and the progression of the cell cycle. Over-expression of D1, due to translocation (t) (11;14), is known to be an initial oncogenic event in mantle cell lymphomas. The abnormal activation of cyclins is responsible for deregulation of the entry of tumour cells into the S-phase and thus genomic and chromosomal instability, which can lead to the appearance of secondary oncogenic events favouring tumour progression or tachyphylaxis. The overexpression of cyclin D1identified in HCL is not identified in HCL-v, however, another activating mutation has been identified in HCL-v; that of CCND3[12]. This mutation leads to a loss of the PEST domain, which controls protein degradation, and thus increases the expression of cyclin D3 [23]. CCND3 mutations have been identified in splenic diffuse red pulp B-cell lymphoma (SDRPL) and Burkitt lymphoma [23, 24]. In these patients, CDK4/ CDK6 inhibitors could be used [24]. TP53 mutations, which are very common in solid tumours, were initially reported in 28% HCL cases and the deletion del(17p13) was reported in 75% of cases [25, 26]. However, these data were not confirmed in more recent series [7, 9-12]. More far-reaching studies are required in order to determine the exact frequency of these anomalies. TP53 mutations are more frequent in HCL-v (Table 1) and are observed in more than 25% cases. The p53 protein is activated in response to various aggressive events, such as DNA damage, oxidative stress, or oncogenic signals. It induces the expression of genes involved in many cellular pathways including cycle arrest, senescence, certain metabolic pathways, and apoptosis. As for cyclins, the inactivation of p53 creates a favourable context for the accumulation of additional oncogenic events. The search for TP53 deletions/mutations is of particular interest in the case of disease refractory to first-line treatment, as alteration of p53 function is known to confer resistance to chemotherapy treatments [27].
NF-kB pathway
The nuclear factor-kB (NF-kB) pathway is essential for the maturation and homeostasis of B lymphocytes as well as the establishment of an effective immune response. In mature B cells, this pathway is primarily activated by the B-cell receptor, various members of the tumour necrosis factor (TNF) family and Toll-like receptors. The NF-kB pathway plays a central role in the activation of hairy cells (Figure 2). Indeed, an in silico study of gene expression in HCL revealed over-expression of the genes regulated by this pathway [28]. KLF2 mutations, always associated with the BRAFV600Emutation, are reported in 10–16% of HCL cases [10, 29, 30]. KLF2 is a transcription factor involved in differentiation, and allows the expression of CD62L, a selectin involved in lymphocyte nodal localisation [31]. KLF2 is also a negative control factor for the NF-kB channel (Fig. 2) [32]. Mutations located near the zinc finger domain or nuclear export signal have also been described in marginal zone lymphoma (MZL) and may affect the transcription factor activity of KLF2, via cytoplasmic relocation of the protein [30]. KLF2 mutations in HCL could thus explain the preferential extra-ganglionic location of the disease and the deregulation of the NF-kB pathway.
Spliceosome
Mutations in the U2AF1 gene have been identified16% of patients with HCL-v [7, 12]. Absent in HCL, these mutations are common in myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML) [33]. U2AF1 is one of the components of the spliceosome that recognises the 3’ splice site end and binds to it. Mutations in U2AF1 are mainly located at two hotspot amino acids (Ser34 and Gln157) in the zinc finger domains of the protein. RNA splicing is altered, resulting in retention of introns or deletion of some or all exons [34]. Identification of these mutations in HCL-v may be of interest by sensitising hairy cells to drugs that modulate splicing [35].
Epigenetics
Epigenetics modifies the function of genes without altering the DNA sequence. There are two types of epigenetic modifications: methylation of CpG islands of DNA and post-translational modification of histones.
DNA methylation
The methylomes of 11 patients with HCL were compared to those of normal B cells and those of patients with CLL or MZL [36]. This analysis confirmed the post-germinative origin of hairy cells. Based on a comparison between methylation and transcriptomic profiles performed in 2004 [21], for half of the genes, an inverse correlation was observed between methylation status and the rate of gene expression. These data suggest that methylation plays an important role. Epigenetic modifications promote constitutive activation of the MAP-kinase pathway, and hypomethylation of the following genes has been demonstrated:
- –IGFR1, which activates signal channels upstream of the pathway;
- –CMKLR1, which encodes a protein that induces phosphorylation of ERK [37];
- –MAP2K1, which encodes MEK1, one of the pathway effectors.
These modifications also modulate the interactions of the hairy cell with its microenvironment. The promoter of the CXCR5 gene, which encodes the CXCL13 chemokine receptor involved in B-cell lymphocyte nodal location, is hypermethylated, which explains the usual absence of lymph node infiltration in HCL [38]. Hypomethylation of FGF2 and FLT3 promoters is also observed. Over-expression of these genes could contribute to the spinal fibrosis present in HCL [39]. FGF2 is responsible for the production of fibronectin [40] by the hairy cell. The FLT3 ligand is involved in binding B cells to fibronectin through the activation of the integrins, VLA-4 and VLA-5, expressed by hairy cells [41]. The methylome interacts with the PRC2 complex (polycomb repressive complex 2) to maintain gene repression. The promoters of two genes encoding for components of PRC2, RBBP4 and SUZ12, are hypomethylated and co-localisation of trimethyl histone markers and CpG island methylation has been observed [42], demonstrating a unique signature of the methylome, characterised by hypomethylation of the BCR-TLR-NF-kB, BRAF-MAPK and cell binding pathways and hypomethylation of cell differentiation markers. It is notable that this profile and signature differs from that observed in MZL.
Histone modifications
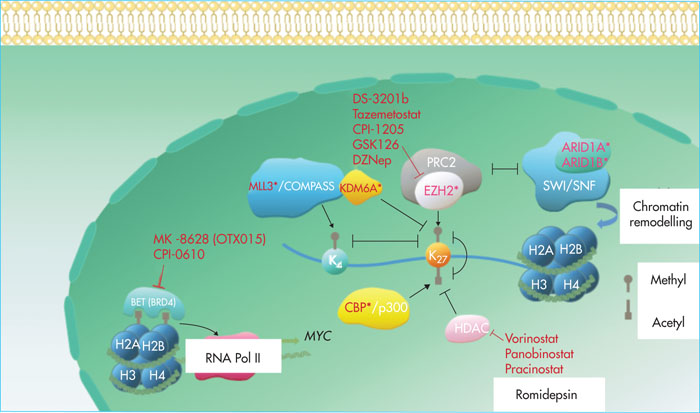
Post-translational modifications of histones regulate the level of chromatin compaction and the accessibility of genes to the transcriptional machinery. The types of modification include methylation, acetylation, phosphorylation and ubiquitinylation. Studies [3, 7, 9-12] have shown a recurrence of mutations in the KMT2C (MLL3), ARID1A, ARID1B, CREBBP, KDM6A (UTX) and EZH2 genes (Figure 3).
Inactivating mutations of KMT2C (MLL3), located at 7q36, have been identified in 15% of patients with HCL. They have also been observed, at the same frequency, in patients with HCL-v [12]. KMT2Cintervenes within the macromolecular complex, COMPASS (complex of proteins associated with Set1), adding mono-, di- or trimethyl tags to lysine 4 of histone 3 (H3K4) (Figure 3). KMT2C is responsible for the monomethylation of H3K4 at transcription activating sequences. The methylation of H3K4 co-locates with the acetylation of H3K27, creating an epigenetic environment, favouring transcription, and limiting methylation of H3K27 by the PRC2 complex [43]. Inactivating mutations of KMT2C are observed in many cancers, including B-cell non-Hodgkin's malignant lymphomas (NHML), cutaneous T-cell lymphomas and LAMs. KMT2C has been identified as a tumour suppressor gene in LAMs associated with a del(7q) deletion. This deletion is often accompanied by NF1 deletion and inactivation of p53. The concomitant presence of these three abnormalities in transplanted haematopoietic stem cells in mice results in the development of LAM. LAMs have a poor prognosis and are resistant to chemotherapy. They may be sensitive to BET inhibitors, which block transcription of the over-expressed MYC oncogene [44]. In 2009, the coactivating role of KMT2C towards p53 was highlighted, and KMT2C was shown to promote the expression of dep53 targets involved in the response to DNA damage [45]. The loss of KMT2C expression leads, in urothelial cancer, to repression of the genes involved in DNA repair by homologous recombination (BRCA1, BRAC2, RAD50, RAD51, etc.). Tumour cells would then be dependent on the alternative system of non-homologous end junctions for DNA damage repair and sensitised to PARP inhibitors [46].
Inactivating mutations of EZH2 (7q36) are rarer and have been observed in two patients with HCL. EZH2 (enhancer of zest homolog 2) is one of the two enzymes of the PRC2 complex, which adds three methyl groups to histone H3 lysine 27 (H3K27) (Figure 3). This allows the PRC1 complex to be recruited, which maintains repression of the target genes, either by chromatin compaction or by direct interaction with the transcriptional machinery at the target gene promoter [47]. In diffuse large B-cell lymphomas (DLBCL) and follicular lymphomas (FLs), mutations in the SET domain of EZH2, leading to increased rates of trimethylation of H3K27, are frequently observed [48, 49]. EZH2 is believed to promote the proliferation and self-renewal of tumoural B lymphocytes. Inactivating mutations of EZH2 are also described in myelodysplastic syndromes [50] and T-acute lymphoblastic leukaemia (ALL). In such cases, inactivation of EZH2 is thought to promote oncogenic activation of the NOTCH1 pathway, which is characteristic of the disease [51]. In the case of EZH2 “gain of function” mutations, EZH2 inhibitors may be used. The first of these to have been synthesised is DZNep (3- deazaneplanocin), an inhibitor of S-adenosylhomocysteine hydrolase, the methyl-donating cofactor of methyltransferases, leading to a non-specific inhibition of histone methylation. Subsequently, more specific drugs, operating in competition with the EZH2 cofactor, S-adenosyl-methionine, emerged, such as GSK126 or EPZ6438 (tazemetostat). The efficacy and safety of tazemetostat is currently being evaluated in several Phase I and II clinical trials in patients with non-Hodgkin's B lymphoma [52].
CREBBP mutations are present in 6% of patients with HCL [12] and approximately 11% of those with HCL-v. In our cohort [10], two patients had a “loss of function” mutation: one with HCL and the other with HCL-v. The CREBBP gene encodes a ubiquitously expressed nuclear phosphoprotein, CBP, belonging to the KAT3 family of histone / lysine protein acetyltransferases. CBP, in combination with p300, is involved in the regulation of many cellular pathways. It modulates gene transcription through the acetylation of lysine 18 and 27 of histone 3 (Figure 3) and by stabilising interactions between transcription complexes with RNA polymerase and additional proteins. CBP is also involved in regulating the cell cycle [53, 54]. Inactivating mutations of CREBBP are frequently found in solid tumours [54]. BecauseCREBBP promotes the activation of tumour suppressor genes, including TP53, it is classically considered itself to be such a gene. In DLBCLs and LFs, CREBBP inactivating mutations may be responsible for an acetylation defect in the BCL6 promoter leading to constitutive activation of this transcription factor. A lack of acetylation of the TP53 promoter is also observed, which decreases its expression [53]. In DLBCL, loss of CREBBP expression sensitises cells to histone deacetylase inhibitors such as vorinostat, which has been approved for the treatment of cutaneous T-cell lymphomas [55]. In ALLs with activating mutations in the KRAS gene, loss of CREBBP expression would promote activation of the RAS/RAF/MEK/ERK pathway without, however, affecting sensitivity to MEK inhibitors [56].
Mutations of ARID1B and ARID1A, most probably inactivating mutations, were observed in four patients with HCL and three with HLC-v. ARID1A and ARID1B are proteins that are part of the ATP-dependent chromatin remodelling complex, SWI/SNF (switch/sucrose non-fermentable). Mutations are mutually exclusive. Subfamilies of chromatin remodelling enzymes catalyse a wide range of chromatin transformations, including movement of histone octamers throughout the DNA and changes in the composition of these octamers and conformation of nucleosomal DNA (Figure 3) [57]. Mutations affecting the different subunits of the SWI/SNF complex are frequently found in cancers. The resulting dysfunction is thought to affect both transcriptional roles, such as the modification of transcription factor binding sites, and non-transcriptional roles, such as the deregulation of DNA repair and chromatin remodelling systems [58]. In ovarian cancer cell lines, loss of function of ARID1A has been shown to result in increased sensitivity to EZH2 inhibitors via inhibition of the PI3K/AKT pathway [59]. Another study investigated the sensitivity of SWI/SNF-deficient cell lines to EZH2 inhibition, the authors demonstrated that the majority of the cell lines were sensitive to EZH2 inhibition by interfering RNAs, but that not all lines were sensitive to EZH2 inhibitors. The observed dependency therefore not only relates to the catalytic activity of EZH2 but also involves the ability of the protein to stabilise the PRC2 complex via its interactions with other proteins of the complex, such as SUZ12. Other types of EZH2 inhibitors that target EZH2 interactions with adjacent proteins [60] need to be developed. Cell lines with RAS oncogene activation are resistant to EZH2 inhibition. Another possible therapeutic approach is to build on the existence of mutually exclusive subunits within the SWI/SNF complex and to target the residual activity of the non-mutated subunit. An antiproliferative effect of ARID1B inhibition has been demonstrated in cell lines with functional deficiency of ARID1A [61]. ARID1B mutations are less frequent than ARID1A mutations. Most of these are inactivating mutations and their functional consequences continue to be under-studied. ARID1B mutations are best known for their role in neurodevelopmental abnormalities such asCoffin-Sirissyndrome. Inactivating KDM6A mutations wereidentified in two of the four HCL-v patients in our cohort [10, 62]. The studies by Dietrich et al.[9] and Weston-Bell et al.[11] reported that one in three patients with HCL was a carrier. One of these mutations occurred after the initiation of treatment with a BRAF inhibitor. In the study by Durham et al. 12], the mutation was detected in 1 in 53 patients with HCL and 1 in 8 patients with HCL-v. KDM6A (lysine demethylase 6A), also known as UTX (ubiquitously tetratricopeptide repeat on chromosome X), is a histone demethylase that targets the di- and trimethyl group of lysine 27 of histone 3 (H3K27) [63]. It is also thought to be a positive regulator of the SWI/SNF chromosome modelling complex [64] and interact with CBP protein [65] and the mixed lineage leukaemia (MLL) complex [66] (Figure 3) [66]. Somatic mutations of KDM6A are frequently reported in cancers [67]. A recent study on multiple myeloma [68] showed that the loss of KDM6A function promotes proliferation, clonogenicity, adhesion, and tumuorgenicity of tumour plasma cells. This loss of function was accompanied by a dependence of the cells on PRC2 complex, sensitising them to EZH2 inhibitors. This dependence has also been shown in a study on bladder cancer [69]. This may be explained by the fact that the PRC2 complex has an antagonistic activity towards KDM6A and thus inhibition of EZH2 may allow the balance between methylation and demethylation to be restored. Approaches using bromodomain BET protein inhibitors [70] or histone deacetylase inhibitors [71] have also been tested on pancreatic cancer cell lines with a KDM6A mutation, with varying efficacy depending on the lines studied. In this type of cancer, the tumour suppressor role of KDM6A is believed to be largely independent of its demethylase activity and is due, in particular, to an increase in MYC transcription. The effect of BET inhibitors is explained by their ability to target MYC[72]. The use of histone deacetylase inhibitors may promote the acetylation of H3K27, which antagonises its methylation by the PRC2 complex.
Epigenetics and therapeutic perspectives
The evidence of epigenetic modifications allows us to envisage the development of targeted and personalised treatments. Vorinostat was the first histone deacetylase inhibitor to be used. Such drugs are now evolving as they are used in combination with one another or in combination with immunotherapies or chemotherapies to overcome resistance and reduce chemotherapy doses. In HCL, these epigenetic alterations are relatively frequent and new therapeutic strategies may be developed based on the concept of synthetic lethality. This concept is the basis for the use of EZH2 inhibitors in various mutational environments. Thus, the presence of an inactivating mutation of KDM6A that encodes a component of the SWI/SNF, MLL3 or CREBBP complex in hairy cells could make them sensitive to such treatments. The rationale lies initially in the antagonistic action of these proteins towards the PRC2 complex. Histone deacetylase inhibitors could be used in the event of CREBBP or KDM6A mutation to restore the balance between acetylation and methylation of H3K27. Finally, BET inhibitors could be used if MLL3 or KDM6A mutations occur. Several of these treatments have been shown to be effective in vitro or in vivo in diseases other than HCL. In vitro studies have yet to be conducted for HCL and HCL-v, after which their clinical use can be considered.
Conclusion
HCL and HCL-v are characterised by the constitutive activation of the MAP-kinase pathway. This constitutive activation is mediated both by activating gene mutations (BRAFV600E) and mutations in the MAP2K1 gene, and by a particular DNA methylation profile. The identifiedmutations are heterogeneous, but some attract attention due to their frequency and/or involvement in the pathophysiology of the disease. Among the recurrent mutations associated with BRAFV600Eare the inactivating mutations of CDKN1B and KLF2. CDK1NB encodes for p27, which negatively regulates the cell cycle and is an antagonist of cyclin D1, which is overexpressed in HCL. Regulation of the cell cycle and senescence thus appears to play an important role in the pathogenesis of the disease. KLF2 encodes a negative regulator of the NF-kB pathway, an important pathway in the development and maintenance of B-cell functions. Mutations affecting epigenetic regulation are recurrent in both HCL and HCL-v. They have an impact upon various enzymes and appear to promote the activity of the PRC2 complex by inactivating its antagonists. The DNA methylation profile also acts synergistically with the PRC2 complex in order to maintain repression of the target genes. Identification of these abnormalities makes it possible to envisage personalised and targeted treatments. One third of HCL patients relapse and those with HCL-v often respond poorly to the various treatments currently available. The use of drugs targeting epigenetic regulators could be promising. The mutational profile of hairy cells is a valuable tool to help distinguish between the different forms of hairy cell proliferation, namely HCL, HCL-v, Splenic Marginal Zone Cell Lymphoma (SMZL) and SDRPL. HCL is characterised by the BRAFV600Emutation and CDKN1B mutation, HCL-v by MAP2K1 mutation, SDRPL by BCOR mutation, and SMZL by mutations in NOTCH1/NOTCH2. Klf2 mutations are present in both HCL and SMZL, and CCND3 mutations in HCL-v and SDRPL.
Liens d’intérêt
Les auteurs déclarent n’avoir aucun lien d’intérêt en rapport avec cet article.