Epileptic Disorders
MENUThe aetiologies of epilepsy Volume 23, issue 1, February 2021

Learning objectives [1]
- •Describe the major aetiologies for epilepsy (i.e. structural, genetic, infectious, metabolic, immune, and neurodegenerative) (L1).
- •Describe the common structural aetiologies (e.g. hippocampal sclerosis, tumours, malformations, vascular lesions, traumatic brain injury, etc.) (L2).
- •Describe the common genetic causes of epilepsy (e.g. monogenic or polygenic inheritance, germline or somatic mutations) (L2).
- •Describe the common infectious causes of epilepsy, including geographical impacts (e.g. bacterial, fungal, viral, parasites) (L2).
- •Describe the common metabolic causes of epilepsy (e.g. inborn errors of metabolism, glucose transport defects, pyridoxine dependent seizures, mitochondrial pathologies) (L2).
- •Describe the common immune causes of epilepsy (e.g. Rasmussen encephalitis, LGI1 antibodies, NMDA antibodies, etc.) (L2).
- •Describe the common neurodegenerative causes of epilepsy (e.g. Alzheimer's disease, Down syndrome, progressive myoclonic epilepsies) (L2).
Major aetiologies of epilepsy
Any brain has the propensity to have seizures, which occur when excitability of an area or areas of the brain exceed a certain threshold. The epilepsies are a group of heterogenous neurological conditions whereby an underlying brain disorder leads to a reduction of the intrinsic seizure threshold, so increasing the propensity for spontaneous recurrent seizures.
The clinician's first diagnostic step is to determine that an event is most likely an epileptic seizure and not one of a range of possible differential diagnoses including dissociative seizures, syncope, parasomnias, movement disorders and other non-epileptic events. The identification of an underlying aetiology is a fundamental subsequent step for the diagnosis and management of the epilepsies. Indeed, since aetiology influences the recurrence risk following a single seizure [2, 3], it can be important in making the diagnosis of epilepsy, for which a seizure recurrence risk of 60% is an accepted criterion [4]. The most recent classification of the epilepsies provides a new framework at three levels and highlights the importance of considering aetiology at each level: seizure type, epilepsy type, and epilepsy syndrome [5]. Aetiological categories in the International League Against Epilepsy (ILAE) classification include structural, genetic, infectious, metabolic, immune, and unknown [5]. In the ILAE curriculum, there is an additional category, neurodegenerative. The aetiologies within neurodegenerative can be placed in other aetiological categories, but, because of the growing clinical importance of neurodegenerative disease, here it is considered separately [6]. These categories are not mutually exclusive; for example, some genetic conditions, such as tuberous sclerosis, cause structural lesions, or inborn errors of metabolism can often be included in both genetic and metabolic aetiological categories. Moreover, there may be multiple risk factors that contribute to the development of epilepsy (e.g. people with a family history of epilepsy have a higher chance of developing epilepsy following a traumatic brain injury).
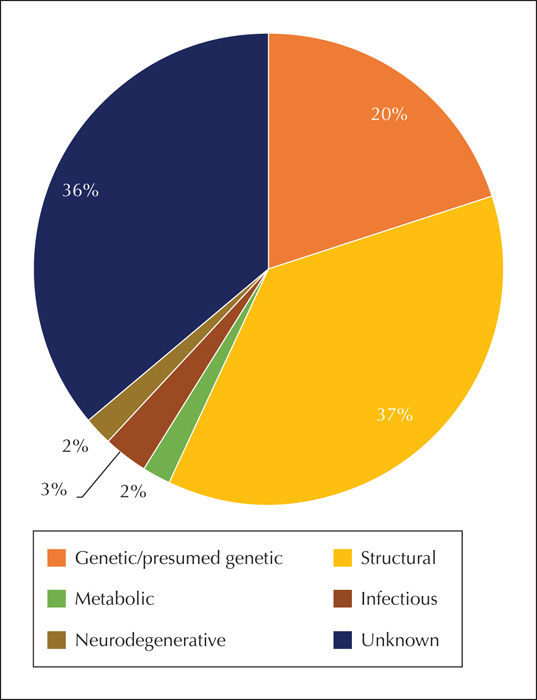
The unknown category rather speaks to our lack of knowledge, or perhaps lack of suitable diagnostic tools, and remains one of the largest categories [7]. As science advances and as diagnostic tools become more widely available, fewer epilepsies will fall into the unknown category, and the aetiological categorisation of certain epilepsies will change. Indeed, recent advances in high-resolution neuroimaging, testing for autoimmune antibodies, and next-generation sequencing (NGS) have revolutionised our ability to identify the aetiology of many epilepsies. Therefore, every patient with epilepsy, in whom the aetiology is unknown, deserves regular review with regard to their underlying aetiology.Although many epidemiological studies investigate the prevalence of each of the aetiological categories (figure 1), they are often performed in resource-rich countries and do not represent the spectrum and prevalence of epilepsy aetiologies globally. Time and place have a considerable impact on the spectrum of aetiologies seen clinically. For example, infectious causes, in particular parasitic infections, are commoner in resource-poor countries. In some endemic communities, neurocysticercosis is the cause of approximately a third of all active epilepsy [8]. Over time, public health measures reduce the incidence of perinatal insults, infectious causes and traumatic brain injury, whilst an ageing population results in an increase in the frequency of stroke and dementia.
Because aetiology plays a central role in the diagnosis and management of people with epilepsy, investigations into cause should be instigated from the outset; for example, neuroimaging, ideally MRI (where available) should be undertaken in all people with a first seizure when a structural aetiology is suspected [2]. The presence of a structural abnormality together with an electro-clinical assessment help to determine prognosis and allow earlier recognition of surgical candidates in those who do not respond to appropriate antiseizure medications [9]. Equally, identification of a genetic cause carries several clinical implications, including focusing further investigations, informing long-term prognosis, and counselling the patient about family risk and prenatal testing options. Moreover, discovering a specific pathogenic gene variant as a cause for epilepsy increasingly allows tailoring of treatment strategies, and targeting of the underlying pathophysiological mechanisms [10]. Infectious aetiologies are the commonest group worldwide and are often associated with structural abnormalities. They also carry specific treatment implications, including surgical treatment [11, 12]. Metabolic causes can refer to acquired metabolic or inborn (genetic) metabolic causes in which there is a specific metabolic defect causing the epilepsy, often there are other clinical manifestations, and biochemical changes reflecting the specific pathway involved [13]. Early diagnosis and treatment are crucial for the prevention of irreversible brain damage [14]. Immune causes refer to epilepsy directly resulting from autoimmune-mediated central nervous system inflammation, causing characteristic clinical features in both children and adults [5]. Also, conditions in this aetiological category often have particular targeted treatments; immunomodulatory therapy is often much more successful than conventional antiseizure medications or epilepsy surgery. Alzheimer's disease and other neurodegenerative disorders are increasingly recognised causes of epilepsy in adults, and their identification has implications for clinical management [15]. Finally, there are still many patients with epilepsy where the aetiology remains unknown. This varies considerably in different regions of the world, depending on access to diagnostic resources.
It is also important to note that specific aetiologies cause a wide range of additional neurological and systemic comorbidities, and can increase the risk of premature mortality. Understanding the breadth of phenotypic manifestations informs the need for comprehensive management strategies [16].
Common structural aetiologies
Any structural lesion affecting the cortex can result in seizures and epilepsy. However, seizure semiology will depend on lesion location and not on the type of lesion. Although neuroimaging can give us an idea of the nature of a structural lesion, definite diagnosis requires histopathological examination, either from resected brain tissue (as part of epilepsy surgery for drug-resistant focal epilepsy) or at post-mortem. All resected brain tissue should undergo a thorough histopathological examination semi-colon protocols for neuropathological workup of epilepsy surgery brain tissue have been specified by the ILAE Commission on Diagnostic Methods [17].
Structural lesions that are commonly resected for the treatment of focal epilepsy can be divided into six main disease categories (table 1): hippocampal sclerosis, brain tumours, malformations of cortical development, vascular malformations, glial scarring (including stroke and traumatic brain injury) and brain inflammation [18].
Hippocampal sclerosis characterised by neuronal cell loss in anatomically defined sectors of the hippocampal formation [19]. The hippocampus becomes atrophic, which is visible as volume loss on MRI T1-weighted sequences and hyperintense signal on T2/fluid attenuated inversion recovery (FLAIR)-weighted sequences [20]. The aetiology of hippocampal sclerosis is likely a complex interplay between genetic background/causes and environmental insults, including prolonged febrile seizures, traumatic brain injury, and infective causes [21]. The ILAE Commission on Diagnostic Methods has proposed a histopathological classification scheme, acknowledging that hippocampal sclerosis is a heterogeneous disorder with distinct patterns of neuronal cell loss that likely result from different aetiologies [19].
A frequent cause of focal-onset seizures are brain tumours, accounting for 10% to 15% of all adult-onset and 0.2% to 6% of all child-onset epilepsies [22, 23]. The broad spectrum of brain tumours were classified by the World Health Organisation (WHO) into four grades (Grade I=benign and IV=malignant), but this has recently been amended, incorporating molecular parameters in addition to histology to define different tumour entities [24]. Brain tumours with a glioneuronal composition are of particular interest in epilepsy, as they often present with seizures in childhood, are usually of low malignancy (Grade I), and occur mostly in the temporal lobe. Typical examples are ganglioglioma and dysembryoplastic neuroepithelial tumours.
Most malformations of cortical development present with drug-resistant seizures in early childhood. Focal cortical dysplasia (FCD) is the commonest epilepsy-associated brain malformation and occurs most often in the frontal lobe. It is classified by the ILAE Commission on Diagnostic Methods into three subtypes: type I with cortical dyslamination, type II with cortical dyslamination and dysmorphic neurons (without or with balloon cells) and type III associated with another principal lesion [25]. Malformations of cortical development are divided into those that result from: abnormalities of neuronal proliferation/apoptosis (such as microcephaly, type II FCD, cortical tubers in patients with tuberous sclerosis complex and hypothalamic hamartomas); abnormal neuronal migration (such as lissencephaly, grey matter heterotopia, double cortex and cobblestone cortex); and abnormal post-migration cortical development (such as polymicrogyria and schizencephaly) [26]. Recent advances in molecular-genetic studies have resulted in specific gene alterations being assigned to many of these malformations (see section on genetic aetiologies below).
Vascular malformations associated with seizures include cortically-located cavernous haemangiomas (cavernoma) and arteriovenous malformations. The vast majority of such lesions are not associated with epilepsy [27], and secondary phenomena, such as intracranial haemorrhage and/or haemosiderin deposition, in surrounding cortex are necessary to generate seizures. Seizure onset is, therefore, usually later compared to other disease categories (table 1). Sturge-Weber syndrome is also associated with seizures; the meningeal angiomatosis can result in chronic hypoxaemia and transient hypoxaemic insults to the underlying neocortex [28].
Glial scarring is another common disease category in focal epilepsies and results from an exogenous brain insult, often traumatic brain injury (usually presenting as glio-mesodermal scar). In patients with early-onset focal onset epilepsy, ischaemic or haemorrhagic stroke are the most common causes for glial scarring (e.g. encephalomalacia after perinatal injury). The epileptogenic mechanisms of gliosis are not yet fully understood. Molecular alterations of astrocytic ion channel composition or increased adenosine kinase production may play a role [29]. Interestingly, glial scars resulting from neurosurgical interventions rarely produce a seizure disorder.
Although less common in surgical series, stroke is among the commonest causes of epilepsy in adults [30, 31]. Acute symptomatic seizures occurring ≤seven days after stroke are defined as early, whilst late seizures (>seven days after stroke) are considered unprovoked [32]. According to the most recent ILAE practical clinical definition, epilepsy can be diagnosed after a single late post-stroke seizure due to the high (>60%) risk of recurrence within the next 10 years [4]. The main risk factors for post-stroke epilepsy following ischaemic stroke include cortical involvement, haemorrhage, and early seizures [33], and these variables are included in a validated clinical tool to predict late seizures/epilepsy after ischaemic stroke (the SeLECT score) [34]. Antiseizure treatment is recommended when post-stroke epilepsy is diagnosed [35], and should be chosen according to the individual patient's profile, whilst there is no evidence supporting the administration of antiseizure medications as primary prevention [36]. Even in the absence of a definite stroke, cerebrovascular disease such as leukoaraiosis is a risk factor for the development of epilepsy though the mechanisms are less clear [33].
Less common overall, but a preventable cause of acquired epilepsies is traumatic brain injury. Acute symptomatic seizures occurring ≤seven days after traumatic brain injury are defined as early, while seizures manifesting after seven days post-injury are defined as late. The severity of brain injury is the leading risk factor for post-traumatic epilepsy development [37]. There is also evidence that location of the trauma, i.e. temporal lobe, and early post-traumatic seizures are predictive factors of post-traumatic epilepsy. No effective prophylaxis for epilepsy after traumatic brain injury is available [38].
Genetic causes of epilepsy
With the revolution in next-generation sequencing (NGS), many genes have been identified that contribute to the aetiology of epilepsy. Genetic variants contributing to epilepsy aetiology can be common or rare, defined by whether they are present in ≥1% or <1% of the population, respectively. There is an inverse relationship between the frequency of a genetic risk factor in the population and its effect size in causing disease [39].
The first genetic aetiologies identified were in familial monogenic diseases, where pathogenic variants were located in the protein coding sequence causing amino acid substitutions (such as a missense variant) or protein truncation (such as nonsense, frameshift, deletion, or splicing variants). Showing that the pathogenic variant has an effect on the function of the encoded protein which can directly impact brain activity or development supports a causal role for the variant. Many genes implicated in monogenic epilepsies follow autosomal dominant inheritance, although more rarely, some follow autosomal recessive or X-linked inheritance. Monogenic causative pathogenic variants are usually rare or unique variants (i.e., rare or absent in healthy populations) and have been identified in a large number of epilepsy genes. In the monogenic epilepsies, phenotype-genotype correlation can be challenging. Even with conditions in which there is a clearly defined genotype-phenotype correlation, such as the association of Dravet syndrome with SCN1A mutations, understanding the inter-relationship remains complex. For instance, several other genes have been implicated in causing Dravet syndrome [40]. Conversely, there are a broad range of epilepsies associated with SCN1A mutations, including mild disorders such as genetic epilepsy with febrile seizures plus [41], and developmental and epileptic encephalopathies (DEEs) which are more severe than Dravet syndrome [42-44].
Many DEEs have a monogenic cause, with the pathogenic variant most frequently arising de novo in the patient (i.e., mutations that arise spontaneously during cell division) [45]. Most patients have de novo dominant mutations, though X-linked and autosomal recessive causes also exist. Identification of the pathogenic variant is critical as a number are potentially treatable, and identifying the underlying disorder can help direct pharmacotherapy or the need for surgical evaluation [46]. The nature of the pathogenic variant and the genetic background influence phenotypic expression. To date, about 50% of patients with DEEs have a cause identified. This may carry crucial reproductive implications for parents and siblings.
Only a minority of epilepsies overall has a monogenic aetiology. Common epilepsies, such as the genetic generalized epilepsies and focal epilepsies, follow complex inheritance. This means they have a polygenic basis, where multiple gene variants contribute to the disorder, with or without an effect from environmental factors. Each variant may have a weak effect size, but when combined their interaction results in epilepsy. Little is understood how these variants combine to reduce seizure threshold and contribute to the pathogenesis of epilepsy.
In the search for common variants causing common epilepsies, the most comprehensive genome-wide association mega-analysis of epilepsies compared more than 15,000 patients with 29,000 controls and identified 16 genome-wide significant loci [47]. These loci, however, do not provide a mechanistic understanding but merely identify genomic regions associated with disease. The alternative approach is to look for rare variants causing epilepsy. A whole-exome sequencing study of 9,000 individuals with epilepsy identified an excess of ultra-rare, deleterious variants in patients, compared with controls [48]. It showed a convergence of the genes implicated in both the common epilepsies and the DEEs, particularly highlighting a ubiquitous role for GABAergic inhibition.
Genetic variants are classified into five different categories: benign, likely benign, variant of unknown significance (VUS), likely pathogenic, and pathogenic, according to the American College of Medical Genetics (ACMG) guidelines [49]. This classification is based on variant type (whether it was previously reported in individuals with disease), segregation in families, de novo occurrence, prevalence in controls, results of functional studies, predicted effect on the protein, location in functional domains, and conservation across species.
An increasingly important issue in the genetics of the epilepsies is the role of somatic mosaicism. Genetic variants are usually germline and present in every cell in the body. If an individual has a germline pathogenic variant, then the variant may be transmitted to their progeny. Mosaicism arises when an individual has two populations of cells; one wild-type (or normal) and one pathogenic. The pathogenic variant may be confined to one tissue (somatic mutation), where it is unlikely to be transmitted to the patient's offspring, unless the patient has gonadal mosaicism. In the DEEs, where most patients have de novo mutations, 8% of patients have a parent with mosaicism [50]. If the percentage mosaicism is low (<13%) in parental blood-derived or salivary DNA, the parent is typically unaffected, whereas, with a higher percentage of mosaicism, the parent may be mildly affected with, for example, febrile seizures. The issue of mosaicism is crucial as it significantly increases the recurrence risk for the family of having a second child with a DEE.
Somatic mosaicism also underlies many malformations of cortical development [51]. Specifically, single germline or somatic activating mutations of mammalian target of rapamycin (mTOR) pathway genes are a major cause of focal epilepsies, with and without brain malformations [52]. These include hemimegalencephaly, tuberous sclerosis complex and FCD [53]. Considerable attention has been paid to FCD IIA and IIB, where germline pathogenic variants can combine with a somatic ‘second hit’ to result in the epileptogenic lesion [54].
In addition to single gene variants, epilepsy can also be caused by chromosomal imbalances, such as deletions or duplications larger than a kilobase (copy number variants, CNV), and chromosomal rearrangements. These often cause complex syndromes with epilepsy, dysmorphic features and additional features depending on the size of the CNV and the gene or genes involved in the CNV. Chromosomal microarray analysis and whole-genome sequencing can detect CNVs. Karyotyping is still required for diagnosis of rare forms of epilepsy, such as ring chromosome 20 syndrome [55].
Identification of a genetic aetiology in epilepsy guides management. The concept of precision medicine implies that the treatment is tailored to reversing the functional alteration caused by the genetic mutation: for example, the use of the ketogenic diet in GLUT1-deficiency syndrome to provide an alternative fuel source to the brain; avoidance of sodium channel blockers in Dravet syndrome due to SCN1A-loss-of-function pathogenic variants; use of sodium channel blockers in SCN8A gain-of-function epilepsies; use of mTOR pathway inhibitors (e.g., everolimus) in tuberous sclerosis complex -associated epilepsy [10]. Genetic variations can also affect treatment response through pharmacokinetic and pharmacodynamic mechanisms (i.e., polymorphisms in genes encoding drug metabolizing enzymes such as CYP2C9 and CYP2C19 genes) or through the association between certain human leukocyte antigen (HLA) alleles and increased risk of idiosyncratic adverse drug reactions (e.g., HLA-B*15:02-associated Stevens-Johnson syndrome induced by treatment with carbamazepine in Han Chinese and other South Asian ethnic groups) [56]. There is also growing evidence that genetic variation/diagnosis could be helpful in guiding the selection of suitable candidates for invasive intracranial monitoring and resective surgery [57, 58]. Recurrence risk is important for reproductive counselling, and varies according to the inheritance pattern and presence of mosaicism. Furthermore, a higher risk of sudden unexpected death in epilepsy (SUDEP) is present in some genetic epilepsies, for example, sudden death can be associated with sodium channelopathies [59].
Case 1
A 32-year-old man had his first seizure, which was quite prolonged, at the age of four and a half months following a febrile illness. There was evidence of developmental regression from around the age of nine months. Since that time, he continued to have fairly frequent seizures, refractory to antiseizure medications. Over time, he experienced multiple type of seizures, including atypical absences, myoclonic, tonic, generalised tonic-clonic and focal-onset with impaired awareness seizures (the latter with mesial temporal lobe semiology). He suffers from severe intellectual disability. On examination, there is evidence of crouch gait. Video telemetry EEG recording at age 21 years showed diffuse slow activity with widespread and multifocal sharp waves seen during wakefulness and sleep. A total of five tonic-clonic seizures were recorded, all of which occurred from sleep. The ictal EEG showed possible left temporal onset. Brain MRI showed left hippocampal sclerosis.
Genetic testing at age 23 identified a likely pathogenic mutation in the SCN1A gene, predicted to result in aberrant splicing. This case demonstrates a dual aetiology for his epilepsy. He has a specific disease-causing genetic variant in SCN1A associated with typical features of Dravet syndrome (i.e., epilepsy onset with prolonged seizures before the age of one year, prolonged febrile seizures, developmental regression after seizure onset, and epileptic encephalopathy with multiple seizure types). He also has a structural abnormality (i.e., left hippocampal sclerosis), in keeping with some of the electroclinical findings, suggesting left mesial temporal lobe onset for his focal-onset seizures.
Identifying both aetiologies was crucial in his case for management, as the genetic diagnosis led to more rational treatment (i.e., withdrawal of sodium channel blockers), and surgical treatment, despite being commonly considered for temporal lobe epilepsy, was ruled out due to the genetic abnormality and related widespread neurological impairment.
Infectious causes of seizures and epilepsy
Severe systemic infections, even if they do not directly affect the brain, can predispose to seizures though pyrexia, the release of cytokines, metabolic dysfunction, and triggering of autoimmunity [11]. Here we will focus on cerebral infections. Cerebral infections caused by bacteria, viruses, fungi and parasites are among the commonest causes of seizures and epilepsy worldwide and are particularly prevalent in developing countries. They can cause seizures via several different mechanisms including the direct effects of infection and damage to brain tissue, the production of toxins by the organism, and the induction of inflammation. Importantly, seizures are an independent risk factor for mortality in patients with cerebral infections. For example, the risk of death in bacterial meningitis was ∼18-fold higher in those with associated seizures [60]. Therefore, infectious causes of seizures constitute medical emergencies in many circumstances and prompt treatment should be directed both at the infectious agent and at the seizures.
In Sub-Saharan Africa, infections are the cause of epilepsy in up to 26% of patients [61]. The commonest associations in adults with convulsive epilepsy are malaria or fever (OR = 2.28), Toxocara Canis (OR = 1.74), Toxoplasma Gondii (OR = 1.39), Onchocerca volvulus (OR = 2.23), and Taenia Solium (OR = 7.03) [62]. Among parasitic brain infections associated with epilepsy, neurocysticercosis is the most common worldwide, and, in endemic areas, up to 30% of patients with seizures in the community have neurocysticercosis [63].
Acute seizures are common in all types of viral encephalitis and the risk of epilepsy depends on the type of virus and the occurrence of early seizures. The commonest identified cause of sporadic viral encephalitis is herpes simplex virus (HSV) type 1; other important causes include HSV type 2, cytomegalovirus, varicella zoster, and enteroviruses. Endemic encephalitis is often the result of arthropod-borne viruses, which show specific regional distributions, and include Japanese B encephalitis, West Nile virus, and Nipah virus [64].
Nearly one million people develop bacterial meningitis worldwide annually, often caused by Haemophilus Influenzae, Streptococcus Pneumoniae and Meningococcus. Bacterial meningitis is much more common in developing countries and carries a high risk of neurological sequelae. In developing countries, Tuberculous meningitis (often associated with HIV) is an important cause of seizures and epilepsy, which occur in up to 15% of patients [61]. This contrasts with about 3% of people with HIV who have new-onset seizures, mostly related to toxic or metabolic causes [65].
In a classic epidemiological study of encephalitis and bacterial meningitis in the United States [66], analysing 8,767 person-years, acute symptomatic seizures occurred in 44% of patients with encephalitis and 19% of those with bacterial meningitis. Seizures were focal in 74% of patients and bilateral tonic-clonic or unknown onset in the remainder. The overall risk ratio (RR) for developing epilepsy as compared to those without a central nervous system infection was 6.9, and this was higher in the first four years after infection (RR=10.8) than after four years (RR= 4 to 5). The risk of epilepsy was much higher in patients with encephalitis (RR=16.2) than meningitis (RR=4.2), and the presence of acute seizures was a strong predictor of developing epilepsy (table 2).
Infectious causes of epilepsy have important implications for treatment because of complex drug interactions. For example, enzyme-inducing antiseizure medications can reduce serum levels of antiretroviral and anti-helminthic drugs, and rifampicin and meropenem decrease serum levels of some antiseizure medications. Prevention and treatment of infection aetiologies may also reduce the risk of seizure recurrence and development of epilepsy. There is some evidence from treatment trials of neurocysticercosis that treatment with anthelminthic drugs significantly reduces the burden of seizures [67].
Case 2
A 31-year-old female engineer working in Africa developed a flu-like illness with myalgia and fever. She was prescribed anti-malarial medications. Seven days later, she developed bilateral tonic-clonic seizures which progressed to convulsive status epilepticus. Therapy with phenobarbital, valproate, phenytoin and levetiracetam were ineffective. She was transferred to a tertiary care centre. She was intubated and put in a therapeutic coma. MRI revealed a high signal in both mesial temporal regions. Serum was positive for herpes simplex virus-2, which is an uncommon cause of encephalitis in adults as herpes simplex virus-1 is more common. Extensive testing for other infectious and autoimmune causes was negative. She was treated with antivirals and broad-spectrum antibiotics. EEG demonstrated multifocal discharges and seizures. Eventually, she emerged from status epilepticus after two weeks. One year later, she remains with drug-resistant, multifocal epilepsy with frequent focal impaired awareness and focal to bilateral tonic-clonic seizures, as well as cognitive decline and depression.
This case illustrates a number of important lessons. Infective causes can be difficult to diagnose, and there must be a low threshold for considering treatable infectious causes of epilepsy. Although seizures were treated early, the presumed infectious aetiology was not treated for almost one week. This delay was probably associated with more extensive damage, a worse neurological outcome, and a more severe epilepsy.
The aetiology of the epilepsy here is infectious but the epilepsy is due to the consequent damage and so the aetiology is also structural.
Metabolic causes of epilepsy
Metabolic causes of seizures and epilepsy can be either acquired or genetic (inborn). The acquired metabolic causes of seizures can occur through the failure of an organ (e.g. liver, kidney or pancreas), nutritional deficiencies, autoimmune causes (e.g. type I diabetes mellitus, autoimmune cerebral folate deficiency) or exogenous drugs and toxins [68]. Many of these result in acute seizures (often with an acute encephalopathy) rather than epilepsy unless they cause permanent damage to the brain which may occur, for example, with hypoglycaemia or hyperammonaemia. Of the exogenous toxins, alcohol is amongst the commonest causes of seizures in young adults [30]. Although usually associated with acute seizures (especially related to alcohol withdrawal), alcohol is also associated with the development of epilepsy in a dose-dependent manner [69]; the mechanisms underlying this association are unclear but may not be directly related to alcohol consumption but rather comorbidities such as traumatic brain injury and cerebrovascular disease [69].
Inborn errors of metabolism are a rare cause of epilepsy, but conversely, epilepsy and seizures are commonly associated with inborn errors of metabolism (there are over 200 genetic metabolic disorders associated with epilepsy) [70]. Although rare, their importance lies in the fact that the epilepsy often responds poorly to antiseizure medication but well to correction of the metabolic deficit. The inborn errors of metabolism could be considered a genetic cause of epilepsy, however, it is not the defective protein that causes the epilepsy but rather the consequent metabolic abnormality. Broadly, the seizures can result from failure of brain metabolism, vitamin/co-factor deficiency, accumulation of toxins, accumulation of abnormal storage material, disruption of neurotransmitter systems, or associated malformations of cortical development [13, 14, 71].
Inborn errors of metabolism can present at any age but predominantly present in early childhood. There are certain features which should raise suspicion including unexplained neonatal seizures, early myoclonic encephalopathy, recurrent episodes of encephalopathy, developmental delay, progressive neurological deterioration, seizures related to food intake/fasting, recurrent episodes of vomiting, organomegaly, dysmorphic features, ophthalmological abnormalities, progressive myoclonic epilepsy, family history of similar illnesses (and also family history of neonatal death) and parental consanguinity [14, 70].
Here, we will discuss a few of the seizure-related inborn errors of metabolism that are important to recognize because of their treatment implications. Pyridoxine-dependent epilepsy should be considered in two situations: drug-resistant seizures within the first few days of life, or, less commonly, intractable seizures presenting before the age of three years [72], especially if associated with episodes of status epilepticus. The main cause is a mutation in the antiquitin gene (ALDH7A1), which encodes alpha-aminoadipic semialdehyde dehydrogenase – a key enzyme in the lysine catabolic pathway. Mutations in this enzyme result in the build-up of two metabolites – alpha-aminoadipic semialdehyde and piperideine-6-carboxylate. The latter of these binds to and reduces the levels of pyridoxal 5′-phosphate (an enzyme cofactor essential for normal metabolism of neurotransmitters). Supplementation with pyridoxine increases the production of pyridoxal 5′-phosphate and so redresses this deficit. A lysine-restricted diet, the aim of which is to restrict formation of potentially toxic intermediate metabolites, has been recently proposed as an adjunctive treatment. Some patients with the early life form do not fully respond to pyridoxine, in which case addition of folinic acid may be necessary. Low levels of pyridoxal 5′-phosphate can also result from pyridoxine 5-phosphate oxidase (PNPO) deficiency, an enzyme necessary for the formation of pyridoxal 5′-phosphate from pyridoxine [70]. These patients require supplementation with pyridoxal 5′-phosphate.
In the first few months of life, epileptic infantile spasms, especially in association with ataxia, alopecia and dermatitis, can result from biotinidase or holocarboxylase synthase deficiency, both of which respond to supplementation with biotin [70].
Later in infancy and early childhood, mutations in SLC2A1, the gene encoding the glucose transporter, GLUT1, can result in seizures, a movement disorder (in particular paroxysmal exercise-induced dyskinesia) and learning difficulty. The spectrum of seizure disorders caused by SLC2A1 mutations is broad, depending upon the severity of the deficit [73]; this ranges from an early epileptic encephalopathy to myoclonic-astatic seizures, to early-onset idiopathic (genetic) generalised epilepsies (IGEs). Indeed, mutations in SLC2A1 are associated with about 1% of all IGEs; these are often resistant to antiseizure medication. GLUT1 facilitates the transport of glucose into the central nervous system and glia and deficiencies result in low cerebral levels of glucose. Treatment is aimed at providing an alternative energy source for the brain such as ketones from the ketogenic diet.
Urea cycle deficits can result in the accumulation of ammonia and have a variable clinical presentation depending upon the severity of the deficit [74]. The commonest, ornithine transcarbamylase deficiency, is X-linked, resulting in an early, potentially fatal, metabolic encephalopathy in males but a much milder phenotype in females. The seizures and encephalopathy (often along with nausea and vomiting) are precipitated by high protein loads or hypercatabolic states, which occur, for example, with infections. Treatment is dietary and drugs are aimed at reducing circulating ammonia.
Lastly, mitochondrial disorders are commonly associated with seizures and epilepsy as part of the phenotype [70]. These are often multisystem disorders. Typical presentations are mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) and myoclonic epilepsy with ragged red fibres (MERRF) syndromes. Mutations in the mitochondrial DNA polymerase gamma gene (POLG1) are typically associated with Alpers syndrome, characterised by psychomotor regression, seizures and liver disease, but can also be associated with other phenotypes. Status epilepticus in these conditions can often be associated with cognitive and neurological deterioration. Importantly, use of sodium valproate in these conditions can lead to fatal hepatotoxicity [75].
Immune causes of epilepsy
The immune system is divided into a non-specific immune response, termed innate immunity, and a pathogen-specific immune response, termed adaptive immunity. There is increasing evidence that innate immunity through the release of certain cytokines, such as interleukin-1β, tumour necrosis factor-α, and transforming growth factor-β, can play an important role in the development and maintenance of epilepsy in a large range of pathologies [76], including hippocampal sclerosis and FCD. Targeting these cytokines could thus have an anti-epileptogenic effect.
A critical component of adaptive immunity is the recognition of self (immune tolerance) and differentiation of self from foreign material. Adaptive immunity relies on a specific response to foreign proteins through either a directed cellular attack (by T-cells) or production of specific antibodies (by B-cells). The safeguards in this system to maintain immune tolerance can fail, resulting in autoimmunity [77]. Autoimmune disease (mostly type 1 diabetes, Graves’ disease or rheumatoid arthritis) occurs in about 3-5% of the population but it is relatively rare in the brain, probably because of immune privilege (i.e. the immune system has limited access to the brain). Seizures have long been recognized to be associated with certain autoimmune diseases, such as systemic lupus erythematosus, sarcoidosis, coeliac disease, Behcet's, and Hashimoto's encephalopathy [78]. The mechanisms of many of these associations are not clear and range from vasculitis to metabolic derangement. Autoimmune-associated seizures have also been described in paraneoplastic syndromes.
There are, however, an increasing number of autoantibodies specifically associated with seizures [79]. The autoantibodies are directed either against cell surface or intracellular antigens; the pathogenesis of these two groups is very different. These antibodies have varying association with tumours. For example, ovarian cancer is associated with NMDA receptor antibodies, small cell lung cancer is associated with AMPA receptor and GABA(B) receptor antibodies and Hodgkin lymphoma is associated with mGluR5 antibodies. The features of autoimmune encephalitis with surface-directed antibodies are detailed in table 3.
These autoantibody-related seizures respond well to immunotherapy such as steroids, plasma exchange and intravenous immunoglobulins. These antibodies either cause disease by directly affecting the target proteins and/or by fixing complement and mediating an inflammatory reaction. These autoantibody-mediated syndromes result in seizures but rarely result in epilepsy, as the seizures usually resolve once the antibodies have been successfully treated. The exceptions are GABA(A)R and LGI1 antibodies which can result in chronic epilepsy.
This contrasts with seizures associated with antibodies directed against intracellular antigens such as the paraneoplastic antibodies (anti-Hu, Ma, CRMP2 and amphiphysin antibodies) and anti-GAD65 antibodies. The pathophysiology associated with these involves cytotoxic T cells which respond poorly to immunosuppression [80]. Epilepsy commonly follows seizures associated with these antibodies.
Autoantibodies detailed above can also be associated with new-onset refractory status epilepticus (NORSE), but often the cause cannot be identified (possibly due to, as yet, unidentified autoantibodies or possibly even infectious agents) [81]. The prognosis of NORSE is often poor and there is a high incidence of chronic epilepsy in those who survive. In children, there is a particularly virulent form of NORSE termed febrile infection-related epilepsy syndrome (FIRES), the aetiologies of which are still unclear.
Autoantibodies can also be found in people with chronic epilepsy without an obvious acute or subacute syndrome [82]. The pathogenic significance of these antibodies is unclear and may relate to neuronal damage and the exposure of antigens to the immune system or to antiseizure treatment.
The other prominent cause of an immune epilepsy is Rasmussen's encephalitis [83]. Rasmussen's encephalitis is a unilateral inflammation of the cortex, resulting in refractory epilepsy and progressive neurological deterioration, usually characterised by a progressive hemiparesis and cognitive decline. Histopathology during active disease demonstrates discrete T lymphocytic nodules with microglia and perivascular cuffing, and neuronal death. The T cells are cytotoxic T cells that are CD8 positive, indicating adaptive immunity, and release granzyme B. This points to cytotoxic T cell-mediated pathology in response either to a neuronal pathogen or as an autoimmune response. Autoantibodies have also been described in Rasmussen's encephalitis, although it is unlikely that these are pathogenic but rather have occurred because of neuronal tissue destruction. The course of the condition depends upon age; children usually have an aggressive form of the disease with rapid neurological deterioration, whilst adults tend to have a more indolent form. The treatment has conventionally been immunosuppression usually with steroids, plasma exchange and/or intravenous immunoglobulin. Therapies that target T cell function have also been proposed, including tacrolimus and natalizumab.
Common neurodegenerative causes of epilepsy
Epilepsy commonly occurs in people with neurodegenerative disease. However, it is not always clear whether this association is coincidental, the result of the underlying neurodegenerative pathophysiological process, or simply secondary to neuronal death leading to disruption of networks. Epidemiological studies have shown an increased risk of epilepsy in Alzheimer's disease [84] and Parkinson's disease [85]. Nevertheless, the ILAE classification of epilepsy does not specify neurodegeneration as a separate aetiological category, preferring instead to classify the epilepsy under different aetiological categories. However, since the underlying pathophysiologies of the majority of neurodegenerative diseases are unclear, we have included neurodegenerative disease as a separate aetiological category. With improved diagnostic tools and neurobiological understanding, our ability to classify the aetiology of epilepsy in individual patients with neurodegenerative disease will hopefully improve.
The most common neurodegenerative disorders are the dementias, the most common of which is Alzheimer's disease. Alzheimer's disease is typically characterised initially by impaired episodic memory, followed by difficulties in other cognitive domains. Rare early-onset familial Alzheimer's disease has been associated with a number of gene mutations [86], whilst the aetiology of sporadic Alzheimer's disease (98% of all Alzheimer's disease) is unknown. Determining whether there is an association of Alzheimer's disease with epilepsy has been confounded by the high incidence of epilepsy in the elderly and the shared association with other conditions such as cerebrovascular disease or traumatic brain injury. However, a large epidemiological study has demonstrated that dementia, in general, and Alzheimer's disease, in particular, increases the risk of epilepsy approximately four-fold, independent of age and comorbidities such as stroke or trauma [87].
Some cases of epilepsy in dementia behave like epilepsies with a structural aetiology. In these patients, there is a temporal association between dementia onset and subsequent epilepsy, and seizures can be semiologically related to areas of regional atrophy. In most cases, however, the epilepsy cannot be attributed to a clear structural abnormality visible on neuroimaging [5].
The mechanisms underlying epilepsy associated with Alzheimer's disease may relate to both cell loss and also increases in network excitability; this hyperexcitability can lead to compensatory increases in inhibition that impact upon cognition [88]. Epilepsy is more common in certain forms of Alzheimer's disease, such as a particular familial form of familial Alzheimer's disease resulting from a mutation in presenilin 2 [89]. In general, early-onset Alzheimer's disease, which is more likely genetic, carries a much higher risk of epilepsy than late-onset Alzheimer's disease [6]. Recent evidence indicates that disruption to amyloid and tau signalling pathways, similar to those that occur in Alzheimer's disease, may contribute to the cognitive impairment in drug-resistant temporal lobe epilepsy, suggesting a two-way interaction between epilepsy and Alzheimer's disease [90].
An example of a neurodegenerative disorder associated with a genetic condition that results in epilepsy is seen in people with Down's syndrome. Down's syndrome is caused by trisomy 21, and epilepsy is seen in approximately 10% of affected individuals. In children, the most common epilepsy is West syndrome, characterized by infantile spasms [91]. In later adulthood, people with Down's syndrome often develop Alzheimer's disease. The risk of epilepsy increases substantially at the age when the onset of dementia is common, and cognitive decline and seizures are strongly associated [92]. A distinct syndrome, late-onset myoclonic epilepsy in Down syndrome (LOMEDS), can occur with tonic-clonic and myoclonic seizures and progressive cognitive and functional decline [93].
Myoclonic seizures also occur in the progressive myoclonus epilepsies; a group of clinically and genetically heterogeneous, inherited neurodegenerative disorders, characterised by myoclonus, epilepsy and progressive neurologic deterioration [94]. Myoclonus onset is typically in late adolescence to adulthood [95]. The disease mechanisms vary, and aetiologically, the epilepsy may be metabolic and/or genetic in most cases. The most well-known example is the autosomal recessive Baltic myoclonic epilepsy (also known as Unverricht-Lundborg disease/EPM1), in which cystatin B-mutations (CSTB) are thought to cause hyperexcitability and disrupted cortical network function. Myoclonic seizures are a very early feature followed by generalized tonic-clonic seizures that may become less prominent with time. The degree of cognitive impairment and psychiatric comorbidities varies. Other examples among the many progressive myoclonus epilepsies include sialidosis (lysosomal storage disorders) and myoclonic epilepsy with ragged red fibres (MERRF). With increasing knowledge of different progressive myoclonic epilepsies, treatments are emerging that target either the genetic or metabolic mechanisms.
Conclusion
The present ILAE classification of epilepsy categorizes the epilepsies on a number of different levels including seizure types, epilepsy types, and epilepsy syndromes with aetiology as a central component of each [5]. This classification paper stresses the importance of determining the aetiology, as it can play a critical role in decisions about the treatment of the epilepsy and the management of the patient. Rapid advances over the last 30 years, especially in genetics and neuroimaging, have had a considerable impact on our understanding of the pathophysiology and aetiology of the epilepsies. Undoubtedly, as technology advances, our ability to identify causal aetiologies will improve and we will no doubt refine our aetiological categories. We will surely find that many of the assumptions that we have made are incorrect but in the words of Francis Bacon, “truth emerges more readily from error than from confusion”.
Supplementary data
Summary didactic slides are available at www.epilepticdisorders.com website.
Acknowledgements and disclosures
Part of this work was undertaken at UCLH/UCL which receives a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme. SB was supported by the Muir Maxwell Trust and Epilepsy Society.
AA, SB, IB, JZ & IES have no disclosures relevant to the content of the present work.
SW's institution received unrestricted educational grants from UCB Pharma, Sunovion and Eisai.
MCW has received speaker's honoraria and/or consultancy fees from Eisai, GW Pharmaceuticals, Marinus, and UCB Pharma outside of the submitted work.
Key points
- •Epilepsy aetiological categories include structural, genetic, infectious, metabolic, immune, neurodegenerative and unknown.
- •Epilepsy aetiological categories are not mutually exclusive.
- •Identification of epilepsy aetiology may have relevant implications for clinical management, treatment, and prognostic information.
- •The main categories of structural aetiology include: hippocampal sclerosis, brain tumours, malformations of cortical development, vascular malformations, glial scarring and brain inflammation.
- •The majority of epilepsies have a polygenic contribution, with multiple gene mutations, each with a weak effect size. Therefore, genetic testing often fails to reveal clear genetic causes for common polygenic epilepsies.
- •Infectious causes of seizures often constitute medical emergencies and prompt treatment should be directed both at the infectious agent and at the seizures; seizures are an independent risk factor for mortality in patients with cerebral infections.
- •Inborn errors of metabolism are commonly associated with seizures and epilepsy, especially in neonates and young children and often the epilepsy responds well to the correction of the metabolic deficit, rather than to antiseizure medication.
- •Whilst seizures in the context of autoimmune encephalitis associated with surface-directed antibodies respond well to immunotherapy and do not tend to result in chronic epilepsy, seizures associated with antibodies directed against intracellular antibodies respond poorly to immunosuppression and commonly progress to chronic epilepsy.
- •In patients with neurodegenerative disorders, structural, genetic, metabolic, and/or other aetiological factors may contribute to the pathophysiology of epilepsy.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License