Epileptic Disorders
MENUSTXBP1 germline mutation and focal cortical dysplasia Volume 23, issue 1, February 2021

This case was presented orally at the 6th annual conference “Epilepsy: from gene to scalpel”, Moscow, 2018. The patient described below is included as part of a sample studied in connection with research into “Early epileptic encephalopathy associated with STXBP1 mutations: Clinical description of nine novel mutation carriers” (DOI: 10.18821/1560-9545-2017-22-4-182-189).
STXBP1-encephalopathy (STXBP1-E) is a common genetic early-onset epileptic disorder. The gene encoding syntaxin-binding protein 1 (STXBP1), predominantly expressed in the brain, plays an important role in the fusion of presynaptic membrane vesicles. In most cases, STXBP1-E is the cause of major neurodevelopmental disabilities and severe epilepsy beginning in neonates, though a wide range of symptoms is on record, ranging from autism to axial hypotonia, together with extrapyramidal and cerebellar signs. Magnetic resonance imaging (MRI) findings may include a thin corpus callosum and abnormal myelination [1]. Following neurosurgical resection in two cases, neither of which involved seizures, focal dysplasia 1a (FCD) was histologically confirmed [2, 3].
Here, we report a patient with epileptic symptoms similar to those of PCDH19 encephalopathy with temporal blurring which is consistent with FCD.
Case study
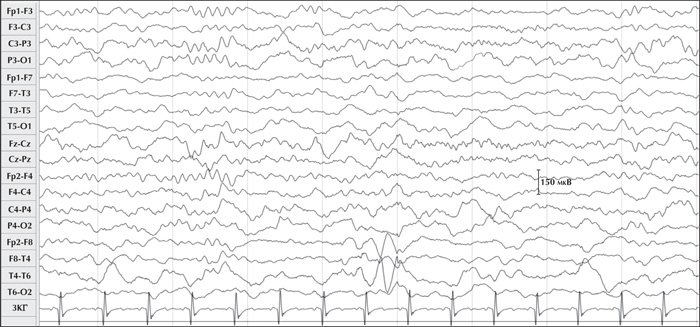
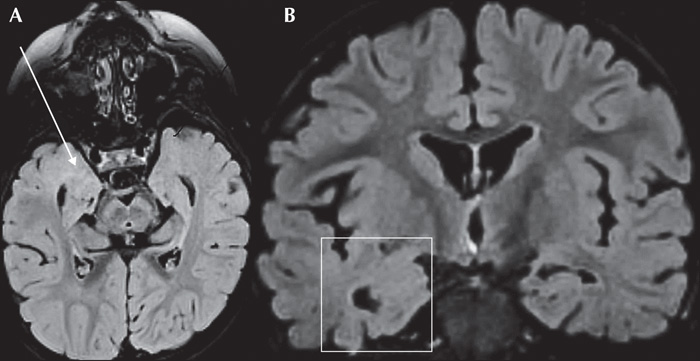
A five-year-old boy, the first child of unrelated healthy parents, developed focal epilepsy at 10 months of age following normal pregnancy and delivery. There was no family history either of any neurodevelopmental disorders or epilepsy. His psychomotor development had been considered normal until his first seizures which were characterized by clusters of brief episodes of behavioural arrest, recurring seven times within three hours. That said, there was no evidence of interictal spikes and indeed the seizures ceased altogether following the administration of 20 mg/kg of valproic acid (VPA). At 14 months, a routine EEG showed background activity which was slow and without spikes, and VPA was withdrawn. At two and a half years, the boy's focal seizures returned with a left tonic head reflex and impaired awareness, followed by secondary generalization, five to six times a day. Slow background activity with right anterior temporal spikes appeared on the EEG (figure 1). On T2 and Flair sequences, 1.5 Tesla brain MRI indicated extensive blurring in the right temporal pole including mesial structures, which were indicative of a dysplastic lesion (figure 2). While the introduction of oxcarbazepine triggered bursts of generalized myoclonus, the seizures stopped altogether for eight months as soon as VPA was re-administered.
Head posturing seizures with impaired awareness recurred (see video), however, and an EEG indicated that both the left and right temporo-occipital areas were affected. Generalized tonic-clonic seizures also occurred, however, it was not possible to pinpoint the focal onset on EEG (supplementary figure 1).
Several AEDs were used in combination with VPA:
- –topiramate which produced myoclonic seizures and deterioration of cognition;
- –levetiracetam which stopped seizures for only two months;
- –both clonazepam and lamotrigine which had a moderate positive effect on the seizures, though the clonazepam proved to be poorly tolerated;
- –rufinamide which had no effect at all;
- –ethosuximide and hydrocortisone which aggravated the seizures;
- –and the ketogenic diet which proved to be ineffective.
The child's parents reported a developmental deterioration: at the age of six, when follow-up had been discontinued, the boy was autistic, suffered from severe cognitive impairment, and was unable to speak. Despite the combination of levetiracetam and lamotrigine, the focal tonic seizures persisted with secondary generalization that affected both sides alternately, several times a week, primarily when he was asleep, with fever occasionally aggravating his condition.
Because the seizures were bilateral, monogenic aetiology was suspected and whole-exome sequencing was carried out using an Illumina NextSeq 500 instrument with an average 97.8x on-target coverage and Nextera Rapid Capture Exome v1.2 reagents for library preparation. A bioinformatics analysis was performed using an in-house software pipeline designed to detect single-nucleotide variants (SNVs). A heterozygous variant in exon 9 of the STXBP1 gene (chr9:130428565G>T, c.784G>T) was identified, leading to replacement of an amino acid at position 262 of the protein domain 3A (p.Asp262Tyr, NM_003165.3). Although this variant is not registered in “1000 genomes”, ESP6500 and ExAC population databases, pathogenicity prediction algorithm AdaBoost considered this missense variant to be pathogenic in all probability (SIFT: 0.000; Polyphen2_HDIV: 1.000; Polyphen2_HVAR: 1.000; MutationTaster: 1.000; PROVEAN: -8.430; LRT: D).
Also, we used Human Splicing Finder tools [4] which showed a possible exonic splicing silencer site and alteration of an exonic splicing enhancer site that may alter splicing.
Sanger sequencing confirmed the variant and its de novo status. This variant was therefore presumed to be pathogenic.
Discussion
Three features distinguish our patient with the p.Asp262Tyr variant from those with the most typical phenotype associated with STXBP1 gene mutation: a) a later epilepsy onset, b) alternating focal seizures and spikes without suppression-bursts or hypsarrhythmia on EEG, and c) features of FCD on MRI.
The variant is a conservative amino acid substitution, p.Asp262Tyr, that occurs at a position that is conserved across different species, and in silico analysis predicts that it may impact secondary protein structure/function. It also leads to potential alteration of splicing, possibly due to the variant being associated with the “GTA” motive. The role of mutations associated with the identified “GTA” motif is significant in gene regulation and may explain the differences compared to the typical picture of STXPB1-related epilepsy [5].
For the vast majority of patients with STXBP1 mutation, epilepsy begins before the age of three months and is suggestive of Ohtahara syndrome in which the brain is extensively affected, due to the suppression-burst pattern, [6, 7].
A few patients with STXBP1 mutation were reported to have Dravet-like syndrome with fever and focal, myoclonic and absence seizures, but with few or no spikes on EEG. For most, MRI was reported to be negative [8]. Focal, brief, alternating temporo-occipital seizures, typically occurring in clusters in infants, are symptoms that are characteristic of PCDH19 encephalopathy [9], a condition most often seen in girls and one that, if the condition occurs in the first year of life, shares many features with those of Dravet syndrome [10]. In such cases, seizures occur in clusters with parieto-occipital discharges. Although seizures are often febrile with affective semiology, they account for no more than half the cases in any one series [11].
The most striking feature of our patient was the MRI of the right temporal lobe which was consistent with FCD. At least four patients with STXBP1 mutation have previously been reported to have MRI and/or histopathological characteristics consistent with those of cortical dysplasia [3, 7]. Only one had an epilepsy phenotype similar to that reported here, namely with alternating clusters of focal seizures, psychomotor retardation, but with no history of suppression-burst or infantile spasms [7].
However, although focal ictal discharges were recorded from both temporal areas in our patient, structural abnormality was seen only on the right side. However, while focal ictal discharges were recorded from both temporal areas in our patient, structural abnormality appeared only on the right side. One explanation could be, in the absence of any structural abnormality, defective inhibition, due to an SCN1A mutation of the sort associated with the Dravet syndrome [11]. However, to date, functional studies have identified only loss-of-function for mutations, including STXBP1 missense mutations [12].
A more likely alternative is that the patient's focal cortical dysplasia was bilateral even though imaging was performed on one side. Even high-resolution 3 Tesla MRI may underestimate the extent of FCD, particularly if it is type 1 FCD [13]. There are only two cases of STXBP1-E with histopathology on record, both of which had type 1 FCD [2, 3]. In both cases, the epilepsy was controlled by means of resection of the whole lesion which confirmed its contribution to the seizure.
MRI carried out before the completion of myelination may fail to detect type 1 FCD because, in the case of the temporal pole, myelination is not normally completed before age four. Indeed, in the case of an 18-month-old child who had undergone an operation, FCD was completely overlooked on MRI which later came to light on histopathological examination [2].
Although repeatedly negative, the last imaging examination was generally carried out before the age of three. When successive MRI provided evidence of abnormal myelination, progressive hypomyelination was mentioned as a possible cause but without any further details [7]. The incidence of FCD in patients with STXBP1 mutation is thus likely to be greatly underestimated, despite the belief that it is a common cause of epilepsy associated with this aetiology.
It is important to note here that in all reported STXBP1 mutation and dysplasia cases, the latter involved the temporal lobes. Since STXBP1 protein appears very early in development, genetic mutations resulting in loss-of-function and syntaxin 1 protein defect could negatively impact the early release of neurotransmitters on which neuroblast migration depends, causing cortical dysplasia, as shown in a mouse model [14]. Since interneurons, which are designed to connect with the temporal lobes, migrate for the longest period, they are the cell types that are the most at risk [15]. In addition, PCDH19, mutation of which is the cause of focal seizures [9], encodes an adhesion protein that also contributes to neuroblast migration. A link between PCDH19 mutation and FCD has also been reported [16]. This finding could explain why seizures associated with the phenotype of our patient shared features with those of PCDH19 encephalopathy [17].
The only patient who underwent molecular genetic analysis based on post-surgical tissue showed somatic STXBP1 mutation in addition to the germline mutation (e.g. tuberous sclerosis) [3]. Because resection of the dysplastic lesion with STXBP1 mutation improved the outcome for only some epileptic patients [2, 3], our case suggests that the risk of relapse from the contralateral side is real and this should thus be kept in mind when considering surgery.
In conclusion, in reviewing the merits of various treatment options for a case of STXBP1 mutation with dysplasia, it seems preferable to try a more conventional treatment such as that for PCDH19 epilepsy before considering surgery [8]. For patients with STXBP1 pathogenic variants, it is our view that, in an effort to identify cortical dysplasia that may have been overlooked, a series of MRI scans should be carried out as soon as temporal myelination has been completed.
Supplementary data
Supplementary figure is available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
We are grateful to Peter Higginson (D.Ed. - University of Massachusetts (Education); prior Director, UNESCO Division of Post-Conflict Educational Reconstruction) who reviewed the manuscript.
None of the authors have any conflict of interest to declare.