Epileptic Disorders
MENUSensitive quantitative detection of somatic mosaic mutation in “double cortex” syndrome Volume 19, issue 4, December 2017


The most frequent recognized cause of subcortical band heterotopia (SBH) is mutation of the doublecortin gene (DCX), which gives rise to an X-linked dominant disorder presenting in females (Gleeson et al., 1998). Rarely, somatic mosaic mutations in the lissencephaly-1 gene (LIS1; also known as PAFAH1B1), affecting only a portion of migrating neurons in the brain, manifest in a “double cortex” pattern of SBH (Jamuar et al., 2014; Mineyko et al., 2010; Pilz et al., 1999; Sicca et al., 2003; Uyanik et al., 2007). Such mutations are estimated to account for 5 to 10% of lissencephaly cases (reviewed in Poduri et al. 2013).
Here, we utilized a sensitive assay to investigate a LIS1 mutation (p.K64X) in a patient with SBH and detect it for the first time in brain tissue, confirming the diagnosis. Using the droplet digital PCR (ddPCR) technique, and two other classic molecular approaches, we also predicted the timing of mutagenesis to be very early during development based on detection of the mosaic mutation in other tissues, a finding consistent with the “double cortex” pattern. We show that the ddPCR technique is powerful for identifying low-level somatic mosaic mutations that cause brain malformations when historical pathological specimens are available.
Materials and methods
Clinical subject
We studied a patient with refractory focal epilepsy associated with mild intellectual disability and delayed speech. Genomic DNA was extracted from blood using the Qiagen QIAamp DNA Maxi Kit (Hilden, Germany). Saliva was obtained using the Oragene kit and genomic DNA was extracted using the prepIT▪L2P kit (DNA Genotek Inc, Ontario, Canada). For formalin-fixed paraffin-embedded (FFPE) brain tissue, phenol-chloroform extraction or the Qiagen FFPE Tissue Kit was used. The Human Research Ethics Committee of Austin Health, Melbourne, Australia, approved this study (Project No. H2007/02961). Informed consent was obtained from the participant for involvement in the study and the use of clinical information and images. All experiments were performed in accordance with the relevant guidelines and regulations of this committee.
Droplet digital PCR
We designed custom probes (WT: VIC-ATTACAAAAGAAGGTAACTAA-MGB-NFQ and K64X: FAM-ATTACAAAAGTAGGTAACTAA-MGB-NFQ) and primers (LIS1.2 FWD-BIOT 5’-TGGAAAAAAAATGGACATCTGTTA and LIS1.2 REV 5’-TGCA GAAGAATGTTATTTTCAGAA) to detect the LIS1 c.190A>T (p.K64X) mutation and wild-type allele. Droplet generation, PCR cycling, and droplet reading were performed according to the manufacturer's recommendations (Bio-Rad, Hercules, CA). Briefly, probes and primers were mixed with 2x ddPCR Supermix for probe (Bio-Rad) at 217 nM and 435 nM final concentrations for each probe and each of the primers, respectively, and mixed with 10 ng of DNA sample to a final volume of 23 μl. In total, 20 μl of reactions were loaded in an 8-channel droplet generator cartridge (Bio-Rad) and droplets were generated with 70 μl of droplet generation oil (Bio-Rad) by using the manual QX200 Droplet Generator. Following droplet generation, samples were manually transferred to a 96-well PCR plate, heat-sealed, and amplified on a C1000 Touch thermal cycler using the following cycling conditions: 95̊C for 10 minutes for one cycle, followed by 40 cycles at 94̊C for 30 seconds and 55̊C for 60 seconds, one cycle at 98̊C for 10 minutes, and 12̊C for an unlimited period. Post-PCR products were read on the QX200 droplet reader (Bio-Rad) and analysed using the QuantaSoft software.
SNaPshot and pyrosequencing assays
The p.K64X mutation was PCR amplified using specific primers to the third coding exon of the LIS1 gene using the reference human gene transcript (RefSeq transcript NM_000430). The standard protocol on a Veriti thermal cycler (Applied Biosystems, Carlsbad, CA) was used for PCR amplification. PCR products were used for SNaPshot and pyrosequencing assays as follows.
For SNaPshot, PCR products were purified using exonuclease I (2 units) and shrimp alkaline phosphatase (5 units) (EXOSAP) treatment. Reactions were then set up using Multiplex Ready Reaction Mix and pooled control and sample PCR products and primers, and thermal cycling completed on a Veriti Thermal Cycler according to the manufacturer's instructions. SNaPshot products were then subjected to post-extension EXOSAP and resolved on a 3730xl DNA Analyzer (Applied Biosystems).
For pyrosequencing, PyroMark Gold Q96 SQA Reagents (Qiagen) and custom biotin-5’ labelled forward primer (LIS1.2 FWD-BIOT 5’-TGGAAAAAAAATGGACATCTGT TA), reverse primer (LIS1.2 REV 5’-TGCAGAAGAATGTTATTTTCAGAA), and sequencing primer (5’-AAAGAAAAAAGACTTAGTTA) were used on a PyroMark Q96 instrument (Qiagen) according to the manufacturer's instructions. Data were analysed with Pyro Q-CpG software (Qiagen, version 1.0.9), as previously described (Lim et al., 2014).
Results
Clinical report
The 40-year-old patient had multiple febrile seizures between 2 and 4 years of age. She had delayed speech and mild intellectual impairment. At age 7 years, focal seizures began, characterized by an aura of fear, followed by loss of awareness and bilateral dystonic posturing. Seizures occurred in clusters and sometimes evolved to bilateral convulsive seizures. She did not respond to antiepileptic drugs and video-EEG monitoring at age 14, in 1991, suggested a left temporal onset. MRI at that time, using a 0.3T instrument, was regarded as normal and she underwent a standard left anterior temporal lobectomy. Histopathological examination of the left temporal lobe, hippocampus, and uncus specimens collected at surgery revealed cerebral neocortex, white matter, and periventricular grey matter. There was mild molecular layer gliosis but no evidence of hippocampal sclerosis or dysplastic cortex. Seizures did not improve post-surgery. Reinvestigation revealed independent bilateral posterior quadrant seizures and 1.5T MRI (figure 1A) revealed bilateral posterior SBH. Clusters of focal seizures continued into her forties despite multiple combinations of antiepileptic drugs.
Mutation detection by droplet digital PCR
ddPCR uses microfluidics and surfactant chemistries to emulsify input DNA into thousands of uniformly-sized droplets, and then amplify them with fluorescently labelled TaqMan probes before measuring fluorescence on a droplet reader, as we and others have previously described (Oxnard et al., 2014; Tsao et al., 2015). Based on fluorescence intensity, the number of mutation positive and wild-type templates is quantified in order to calculate the frequency of a mutant allele. While this approach is “site-specific”, relying on prior knowledge of the precise mutation from pre-screening, it has the advantage of being highly sensitive (down to 0.1% frequency) making it 10-fold more sensitive than sequencing (Abyzov et al., 2017). The approach is highly suitable for old, degraded DNA from FFPE specimens because individual DNA molecules can be investigated. For these reasons, it was the method of choice for the patient studied here.
The patient's formalin-fixed and paraffin-embedded (FFPE) surgical sample from 1991 was retrieved and analysed along with a saliva DNA sample. The brain-derived DNA sample, extracted using the Qiagen FFPE Tissue Kit, was of poor quality (260/280 ratio: 1.95; by Nanodrop), produced a low yield (50 μl at 28 ng/μl by Nanodrop), and was highly degraded given it was obtained and fixed over 25 years ago. Using sensitive ddPCR, the LIS1 p.K64X mutant allele was detected in DNA extracted from FFPE brain tissue using the Qiagen FFPE Tissue Kit (∼5% mutant allele frequency) and saliva using the prepIT▪L2P kit kit (∼13% mutant allele frequency) (figure 1B). No copies of mutant template were detected in the healthy control blood-derived DNA extracted using the Qiagen QIAamp Maxi Kit (figure 1B).
Molecular analyses of other tissues
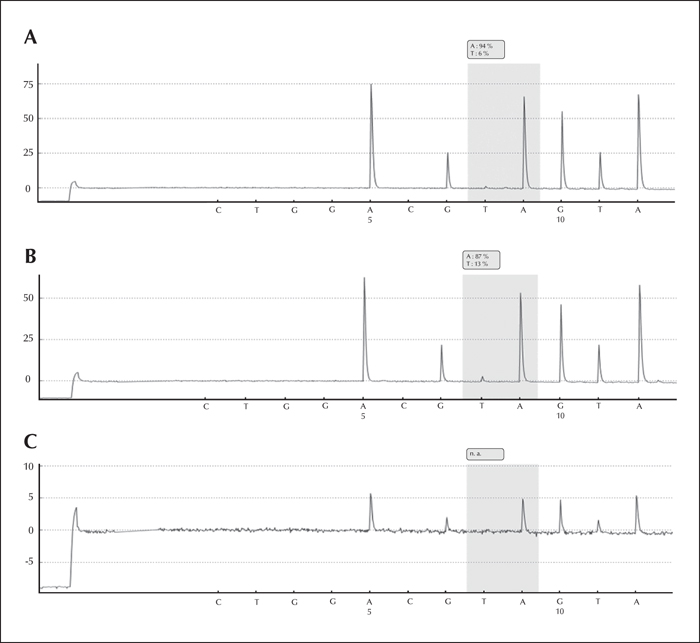
We previously reported the LIS1 p.K64X mutation in blood-derived DNA of the patient by subcloning (Jamuar et al., 2014). This finding was confirmed here by SNaPshot and pyrosequencing assays (∼6% mutant allele frequency) (figure 2A). We were also able to detect mutant allele in saliva (∼13% mutant allele frequency) at the same level as that achieved by ddPCR, providing confirmation by an orthogonal method (figure 2B). However, we failed to identify the allele in phenol-chloroform-extracted brain-derived DNA by pyrosequencing (figure 2C). The presence of the mutation in multiple tissues from different lineages suggests that it arose very early post-zygotically, a phenomenon we have previously described for another epilepsy syndrome (Vadlamudi et al., 2010).
Discussion
Our findings confirm that somatic LIS1 mutation is an important cause of SBH and that such mutations can be present at very low levels in brain tissue due to mosaicism. They may arise very early during development, as suggested by their presence in two distinct cellular lineages. Furthermore, these low-level somatic mosaic mutations have been challenging to detect in FFPE tissue because the DNA quality is generally lower than that from other tissue sources, and the sample often contains more impurities due to the fixing and embedding process which we have shown can interfere with mutation detection methods (Do and Dobrovic, 2012, 2015). As such, the novelty of this report is not the discovery of the mutation itself, in light of our previously published work (Jamuar et al., 2014), but rather the confirmation of the presence of the mutation in the brain by application of a new and highly sensitive technique for low-frequency mutation detection, suitable for old and degraded brain tissue DNA. This is important and timely for patients with brain malformations and epilepsy because much of the biobank tissue available at most clinical research centres is still historical formalin-fixed, paraffin-embedded brain specimens, similar to that studied here. That these historical specimens can be successfully investigated for mutation, even when traditional approaches have failed, is important for future investigation of somatic mutation.
The mutation was not detectable in the archival brain tissue by conventional approaches, such as SNaPshot or pyrosequencing using traditional phenol-chloroform extraction. A ∼5% frequency for the former approach is below the threshold of detection, and for the latter, the poor quality of the FFPE-derived brain DNA sample likely impeded detection. Despite these significant limitations, ddPCR was sensitive enough to detect the low level mutant allele in the brain sample. The use of the Qiagen FFPE Tissue Kit is also likely to have helped overcome these challenges. Together, these approaches are important tools to maximise detection of mutant allele signal from poor quality DNA samples.
The low-level somatic mosaic LIS1 mutation reported here in brain is consistent with the clinical presentation and imaging findings of the patient. Patients heterozygous for LIS1 mutation have the much more severe defect of lissencephaly, presumably a consequence of all neurons expressing the mutant allele, leading to a more diffuse and severe neuronal migration defect (Gleeson et al., 1998). This methodology is likely to have significant utility for a variety of other brain malformation syndromes associated with epilepsy for which there is prior knowledge of the somatic mosaic mutations involved from pre-screening, such as focal cortical dysplasias, or for which there is a known recurrent mutation, such as in Sturge-Weber syndrome (Shirley et al., 2013). In addition, this methodology may be used for important applications in situations where available DNA templates for neurological diagnosis are present at low levels or are of poor quality, such as from FFPE tissue.
Acknowledgements and disclosures
We thank the patient for her participation in this study. Rebekah Stubbs (Epilepsy Research Centre, University of Melbourne) is acknowledged for performing genomic DNA extractions. This study was supported by National Health and Medical Research Council Program Grant 1091593 to SFB, Project Grant 1129054 to SFB, Project Grant 1079058 to MSH, and a RD Wright Career Development Fellowship (1063799) to MSH.
SFB discloses payments from UCB Pharma, Novartis Pharmaceuticals, Sanofi-Aventis, and Jansen Cilag for lectures and educational presentations, and a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies. AD has given lectures and educational presentations for Bio-Rad. The remaining authors have no conflicts of interest to declare.