Epileptic Disorders
MENUResponse to lacosamide monotherapy in a patient with medically refractory Jeavons syndrome: a case report and review of the literature Volume 22, issue 5, October 2020

Jeavons syndrome or eyelid myoclonia with absences (EMA) is a childhood genetic generalized epilepsy which was first reported in 1932, the main features of which were clearly described by Jeavons in 1977 (Striano et al., 2009). It is responsible for 2.7-12.9% of generalized epilepsies and up to 2.7% of all epilepsies (Smith et al., 2018). However, it is frequently under-reported and under-recognized (Smith et al., 2018). Onset is typically between 2-14 years of age with female predominance (Striano et al., 2009; Smith et al., 2018). It is characterized by:
- –eyelid myoclonia (hallmark) with or without brief absences;
- –eye closure-induced seizures or electroencephalographic (EEG) paroxysms (brief generalized polyspikes and/or generalized spike-wave activity at 3-6 Hz, elicited by eyelid closure which may disappear in the dark);
- –and photosensitivity or photoparoxysmal response (eyelid myoclonia provoked by photic stimulation which may disappear when eyes are closed in total darkness) (Striano et al., 2009; Smith et al., 2018). Generalized tonic-clonic seizures may frequently occur (23.3%) and rarely, patients may have myoclonic seizures (Striano et al., 2009; Smith et al., 2018).
Several years after its launch onto the market, the importance of lacosamide in genetic generalized epilepsies including Jeavons syndrome is still uncertain, but pathophysiological mechanisms, along with some case reports and uncontrolled trails, suggest plausible efficacious benefits (Strzelczyk et al., 2018).
To our knowledge, there are no previous reports of lacosamide monotherapy in medically refractory Jeavons syndrome. We describe a patient with medically refractory Jeavons syndrome with highly favorable response to lacosamide monotherapy.
Case study
A 15-year-female presented with uncontrolled absence seizures that initially started at nine months of age. Seizures were characterized by arrest of activity with blank stare, unresponsiveness and eyelid fluttering lasting from 5 to 30 seconds each. She had no history of generalized tonic-clonic seizures. She carried the diagnosis of childhood absence epilepsy since the age of three years.
Her past medical history was significant for attention deficit hyperactivity disorder, oppositional defiant disorder, anxiety, obsessive compulsive disorder and history of head trauma at the age of one year without loss of consciousness. She had normal birth and development and a previous MRI performed at the age of six years was within normal limits.
Between the ages of 3 and 12, she was given adequate trials of ethosuximide, valproic acid, lamotrigine, topiramate and the ketogenic diet, either as monotherapy or in combination without adequate seizure control.
Between the ages of 12 to 14, she remained off anti-seizure drugs because of poor seizure control on medications and medication-related behavior and mood side effects. She continued to have multiple daily absence seizures (up to 25 per day). At the age of 14, she was admitted to the Pediatric Epilepsy Monitoring Unit to clarify the diagnosis of her epileptic syndrome, as childhood absence epilepsy is generally expected to resolve by this age.
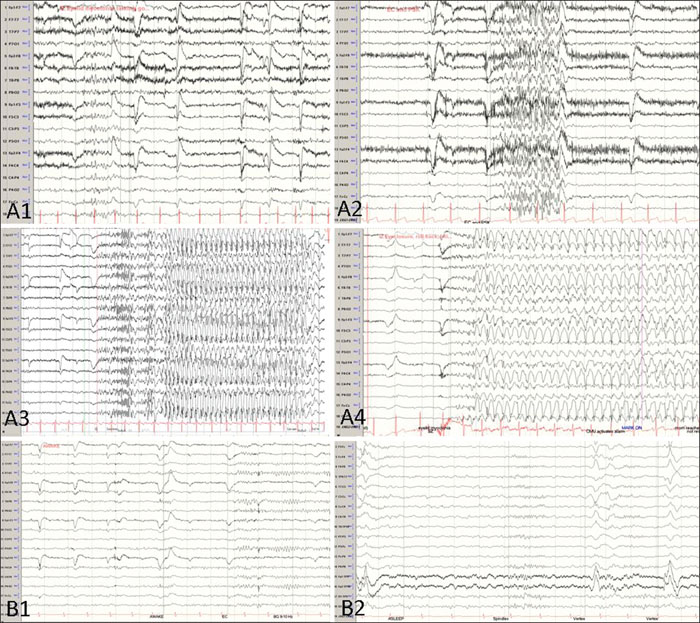
During 24-hour video-EEG (electroencephalography) monitoring, we captured nine clusters of her typical seizures. On EEG, normal awake and sleep background rhythms were interrupted by bursts of generalized polyspikes and generalized spike-wave complexes of 3-4 Hz, most of which were elicited by eyelid closure (EEG paroxysms) and associated with eyelid myoclonia. She was noted to have frequent eyelid myoclonia, some of which evolved into absence seizures, and eyelid closure-induced absence seizures were seen on the EEG characterized by 3-4-Hz spike-wave complexes (figure 1A). These EEG changes associated with eyelid closure were not evident in the dark. Hence, her epilepsy was re-defined as generalized epilepsy with eyelid myoclonia (Jeavons syndrome).
Given her failure of response to multiple medications in the past and her psychiatric history, a trial of lacosamide was decided. She was loaded with 200 mg of lacosamide and started at a maintenance dose of 100 mg twice daily. The EEG showed a dramatic response with resolution of seizures and dramatic improvement in interictal discharges (figure 1B). She remained seizure-free for 11 months on lacosamide monotherapy, after which seizures recurred in the setting of medication non-compliance.
Discussion
Jeavons syndrome is generally a lifelong condition with up to 80% of patients experiencing drug-resistant epilepsy and requiring polypharmacy (Smith et al., 2018). Therefore, it is likely to be managed by both pediatric and adult epileptologists (Smith et al., 2018). Hence, it is a significant syndrome for all neurologists to identify in order to manage appropriately (Smith et al., 2018).
There is limited literature on anti-seizure drug treatment for Jeavons syndome (Parissis et al., 2014; Smith et al., 2018). There have been no randomized controlled trials. Treatment decisions are generally based on anecdotal evidence and case reports (Parissis et al., 2014). Valproic acid, lamotrigine, ethosuximide, levetiracetam, phenobarbital and benzodiazepines are commonly used with inconsistent efficacy, with drug-resistant cases requiring combination therapy (Parissis et al., 2014; Smith et al., 2018). A pilot open-label trial of levetiracetam use as monotherapy or adjunctive therapy demonstrated 80% response rate with acceptable tolerability (Biondi et al., 2008). Lens therapy was deemed effective in up to 75-78% of patients with photoparoxysmal response in two studies, but no studies have been performed specifically for use in Jeavons syndrome patients. Dietary interventions and neuromodulation have also not been thoroughly explored (Smith et al., 2018). Carbamezapine, phenytoin and vigabatrin may worsen seizures (Smith et al., 2018).
A retrospective study at the Mayo clinic showed that even though lacosamide was not commonly used, among the four patients who were tried, two had effective response (50% response rate) although the details of whether this was monotherapy or a combination were not clear (Smith et al., 2018).
Due to its efficacy and favorable side effect profile, several studies (Afra and Adamolekun, 2012; Zangaladze and Skidmore, 2012; Sodemann et al., 2014; Yorns et al., 2014; C. et al., 2015; Strzelczyk et al., 2018) have reported successful treatment of genetic generalized epilepsy including juvenile myoclonic epilepsy (JME), generalized tonic-clonic seizure only, and some refractory cases of absence status epilepticus with lacosamide as monotherapy or an adjunct therapy (table 1 summarizes the response to lacosamide therapy in genetic generalized epilepsy based on various studies in the literature).
Medications that are traditionally used in Jeavons syndrome and other generalized epilepsies are broad-spectrum and it is not known which mechanisms are most responsible for their efficacy in these epileptic syndromes.
Lacosamide exerts its action by selectively enhancing slow inactivation of the sodium channel, unlike traditional sodium channel anti-seizure drugs (Rogawski et al., 2015). It may be possible that the method by which it inactivates sodium channels plays a role in its efficacy as treatment for this syndrome or that it has a mechanism that we are not yet familiar with.
In 2017, the ILAE recognized eyelid myoclonia with epilepsy as a generalized seizure type, as a non-motor (absence) category along with eyelid myoclonia (Fisher et al., 2017). However, studies have shown phenotypic overlap between JME and Jeavons syndrome (Smith et al., 2018). Therefore, Smith et al. suggested that this may be a type of myoclonic epilepsy (Smith et al., 2018). GABA-A and chloride channelopathies have been associated with JME, but not sodium channel abnormalities (Zangaladze and Skidmore, 2012). Based on twin studies, a genetic etiology of Jeavons syndrome has been suggested (Smith et al., 2018). Although, in general, Jeavons syndrome is thought to be caused by polygenic variants with or without environmental influence, monogenic variants and genetic mutations have been reported to manifest with a Jeavons syndrome phenotype, including unique variations in CHD2 (chromo-domain helicase DNA binding protein 2) (Smith et al., 2018), KIAA2022 mutation (Samanta and Willis, 2020), KCNB1 mutation (Marini et al., 2017) and NAA10 mutation (Valentine et al., 2018). Even though no sodium channelopathies to date have been reported in association with Jeavons syndrome, it is interesting to note that sodium channel abnormalities have been reported in some generalized epilepsies phenotypically overlapping with JME (Zangaladze and Skidmore, 2012). Therefore, it may be possible that some patients with Jeavons syndrome could have unknown sodium channelopathies which could explain the efficacy of lacosamide in this syndrome. Unfortunately, our patient was unable to undergo genetic testing due to current medical insurance limitations for genetic testing in older children and adults with epilepsy.
In conclusion, we present a case of medically refractory Jeavons syndrome (EMA) with seizure resolution in response to lacosamide monotherapy at standard daily doses. This highlights the potential role of lacosamide as an option for this syndrome if other drugs are ineffective or not tolerated.
Moreover, given the favorable side effect profile and low teratogenic potential, lacosamide could be considered as an add-on anti-seizure medication or as one of the initial treatment options for Jeavons syndrome in certain patient populations or for those whose co-morbid conditions do not allow the use of other drugs. This case highlights the need for randomized prospective controlled studies with a large sample size to better elucidate efficacy and potential utility of this drug, both as initial or adjunct therapy for this syndrome and other generalized epilepsy syndromes.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Disclosures
Dr Pestana Knight is co-PI for a HRSA grant that is not relevant to this case report. None of the other authors have any conflict of interest to declare.
* This work has previously been presented at American epilepsy society meeting in December 2018. This was presented as a poster.