Epileptic Disorders
MENUProgressive intellectual impairment in children with Encephalopathy related to Status Epilepticus during slow Sleep Volume 21, supplement 1, June 2019

Encephalopathy related to Status Epilepticus during slow Sleep or ESES is an age-dependent and self-limited syndrome whose distinctive features include a characteristic age at onset (with a peak around 4-5 years), heterogeneous seizure types (mostly focal motor or unilateral seizures during sleep and absences or falls while awake), a typical EEG pattern (with continuous and diffuse paroxysms occupying a significant proportion of slow wave sleep) and a variable neuropsychological regression consisting of IQ decrease, reduction of language (as in acquired aphasia or Landau-Kleffner syndrome), disturbance of behaviour and motor impairment (in the form of ataxia, dyspraxia, dystonia or unilateral deficit) (Tassinari et al., 2000). The favourable seizure outcome is independent of the etiology and is observed also in cases with cortical malformations such as multilobar polymicrogyria (Guerrini et al 1998). The characteristic EEG patterns during slow wave sleep also disappear at approximately the same time, but focal interictal spikes may persist (Morikawa et al., 1989; Bureau, 1995).
The first description of sub-clinical electrical status induced by sleep in children dates back to 1971 when Patry, Lyagoubi and Tassinari described in six children a peculiar EEG pattern occurring almost continuously in sleep, characterized by apparently ‘subclinical’ spike and wave for a variable length of time. Tassinari introduced the term “Electrical status epilepticus during slow sleep” and this was originally defined as status epilepticus occurring during at least 85% of slow sleep (Tassinari et al., 1977). More recently, the proportion of affected slow sleep required to affect cognition and behaviour has been recognised to be lower, with some authors suggesting that =>50% can be associated with cognitive sequelae. The most typical paroxysmal discharges on EEG are spike wave at 1.5 to 3.5-Hz polyspikes and polyspikes and wave. Secondary bilateral synchrony is the mechanism underlying continuous spikes and waves during slow sleep. In this respect, the apparently generalized seizures (absences, tonic-clonic seizures) occurring in this condition have, in fact, a focal onset (Tassinari, 1995). ESES can be present at various evolutionary stages within a spectrum of diseases, the prototypes of which are continuous spike and wave in slow sleep (CSWS), Landau Kleffner Syndrome (LKS) and some patients initially presenting with childhood (benign or rolandic) epilepsy with centrotemporal spikes (BECTS) (Galanopolou et al., 2000). Children with ESES generally demonstrate a global neuropsychological disturbance whereas those diagnosed with LKS typically display an isolated language disorder. In both syndromes, recovery of language functions may be associated with disappearance of continuous spike and slow wave during slow wave sleep. The neuropsychological disturbances are suspected to be a function of the genetic or symptomatic origin of the underlying epileptic condition, the cortical area of the primary focal paroxysmal activity, the patient's age and the severity and duration of the EEG abnormalities (De Negri, 1997).
ESES is a rare disorder although accurate incidence is difficult to determine. Morikawa et al found an incidence of 0.5% among 12,854 children evaluated during a 10-year period (Morikawa et al., 1995). The Landau-Kleffner syndrome (LKS) and Encephalopathy related to Status Epilepticus during slow Sleep(ESES) are rare childhood-onset epileptic encephalopathies in which loss of language skills occurs in the context of an epileptiform EEG activated in sleep. Although in LKS the loss of function is limited to language, in ESES there is a wider spectrum of cognitive impairment. The two syndromes are distinct but have some overlap. Whether ESES as a whole should be defined as an independent syndrome or as an electroclinical feature of many epilepsy syndromes is a source of debate (see Hirsch et al., p. S5-S12). The duration of electrical status epilepticus during slow sleep has been correlated with the final neuropsychological outcome (Rousselle & Revol, 1995). It has also been suggested that electrical status epilepticus during slow sleep is a model for prolonged cognitive impairment induced by interictal paroxysmal activity (Tassinari, 1995; see also Tassinari and Rubboli, p. S82-7). “Interictal paroxysmal activity” may interfere with different cognitive processes, as demonstrated by neurophysiological, neuropsychological, and biochemical studies (Binnie, 1993; Wasterlain et al., 1993). It has been suggested that interictal epileptiform discharges have a role to play in the language regression seen in some children with autism, however, other studies have contradicted this view (Ballaban-Gil et al., 1998; De Menezes et al., 1998; Goldberg et al., 1998). What is certain from genetic studies is that autism, ESES and epilepsy may share the same underlying aetiologies (Lesca et al., 2012; Wolff et al., 2017).
Following resolution of ESES, improvement in language dysfunction, learning disability and psychiatric disturbances generally occurs but this is variable and individualized. The majority of affected children never return to normal levels, particularly for verbal and attention abilities (Morikawa et al., 1985; Roulet Perez et al., 1993; see also Arzimanoglou and Cross, p. S71-5). Margari et al followed 25 patients (19 male) from 2 to 16 years of age (mean age: 6 years±3 SD) to examine the presence and course of neuropsychiatric disorder (mean duration of follow-up: 3.9 years) (Margari et al., 2012). At diagnosis of CSWS, 54% of patients had behavioral problems, 37.5% mental retardation, 33% learning disabilities, 17% developmental coordination disorder, 12.5% language disorder, and 8% pervasive developmental disorder. During the follow-up, neuropsychiatric dysfunctions remained unaltered in 52% of the patients, worsened in 24%, and improved in only 24%. The authors suggest that CSWS may be associated with a broad spectrum of neuropsychiatric disorders and may promote their worsening over time. Despite the apparent developmental impact of the ESES, there has been very little published related to neuropsychological outcomes, with only two very small cohort studies, e.g.Seegmüller et al. (2012)n=10 patients, and Scholtz et al (2005)n=7 patients.
The current study aimed to provide data on a larger series of children with treated ESES using standardised neuropsychological tests. We aimed to provide some preliminary data on the impact of ESES on the developing brain in terms of intellectual development in middle to late childhood.
Methods
Participants were identified by reviewing reports of 24-hour ambulatory EEG over a period of five years (n=2200). The recordings of those who exhibited marked sleep accentuation of epileptiform activity were reviewed. Those who fulfilled the diagnostic criteria for ESES, i.e. >85%, were identified. We also included children who had abnormal activity occupying 60-85% of slow wave sleep if there was a history of change in behavior and/or recent loss of skills. Children who were in non-convulsive status with a continuous dysrhythmia, awake and asleep, were excluded. Clinical records were reviewed to determine seizure types, aetiology, and behavioural and EEG features.
This was a retrospective study with cases managed by several clinicians and no uniform or standardized therapeutic paradigm for ESES. Cases were treated with several different anti-epileptic drugs including clobazam, prednisolone and valproate with variability in dose and combination of different treatments. In addition, the sequence and duration of different therapies was not controlled therefore the data on medication use in individual cases is not presented as no valid conclusions can be drawn from this data.
Participants
Twenty-two participants met inclusion criteria (13 males and nine females). The median age of the sample at initial assessment was 6 years (mean: 6.8, sd 2.9 years) with a range of 4-16 years. At follow-up, the median age was 7.5 years (mean: 9.3, sd 3.4 years) with a range of 6-19 years. This created a mean latency period between assessments of 2.5 years. The clinical histories of all patients are described in table 1. Thirteen participants were reassessed within three years and eight participants within 3-5 years. Five of 22 had a significant neonatal history and 5/22 had abnormal imaging using MRI/CT. There were heterogenous epilepsy types (see table 1).
Measures
Participants were assessed using the WISC-III (Wechsler Intelligence Scale for children, version 3, UK norm, UK-edition) or the WPPSI-R (Wechsler Primary and Preschool Scale of Intelligence, revised, UK-edition). When patients were unable to complete all subtests of the WISC/WPPSI, pro-rated scores were derived using statistical techniques outlined within the manual. As this was a retrospective study there was no specified time period between the first and subsequent neuropsychological evaluations.
Results
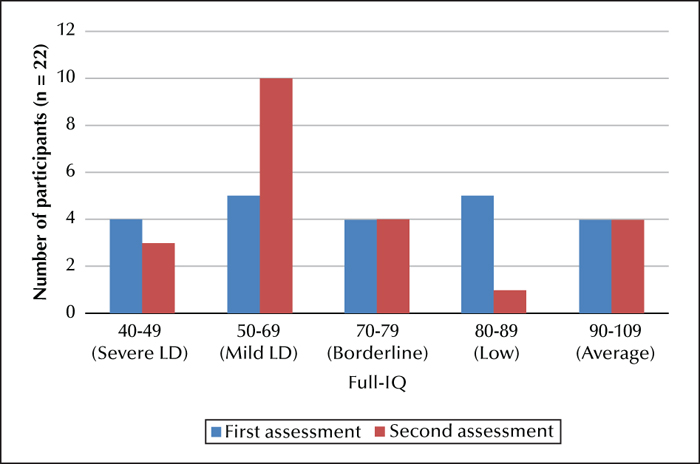
At the time of their initial assessment, nine participants had a measured IQ within the learning disability range, four within the severe range (IQ: 40-49) and five within the mild to moderate range (IQ: 50-69). Four participants had an IQ within the borderline range (IQ: 70-79), five had an IQ within the low-average range (80-89), and a further four had an IQ within the average range (90-109). At follow-up assessment, thirteen participants had measured IQs within the LD range, three within the severe range, and ten within the mild to moderate range. Four participants had an IQ within the borderline range, one within low-average, and four within the average range. Therefore, 41% of the sample had a measured IQ within the learning disability range at first assessment compared to 55% at second assessment; an increase of 14% (figure 1).
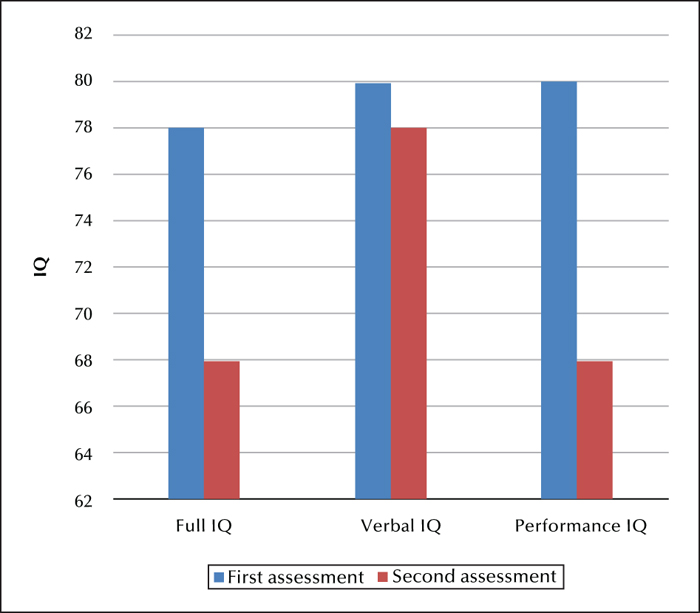
As the data were not normally distributed, a repeated-measures non-parametric analysis (Wilcoxon) was used to compare group median FSIQ, VIQ and PIQ between assessment points (figure 2). There were significant differences in Full-Scale IQ with a small effect size (p=0.042; d=0.3), and performance IQ with a medium effect size (p=0.031; d=0.4). There was no significant difference in VIQ across time (p=0.421).
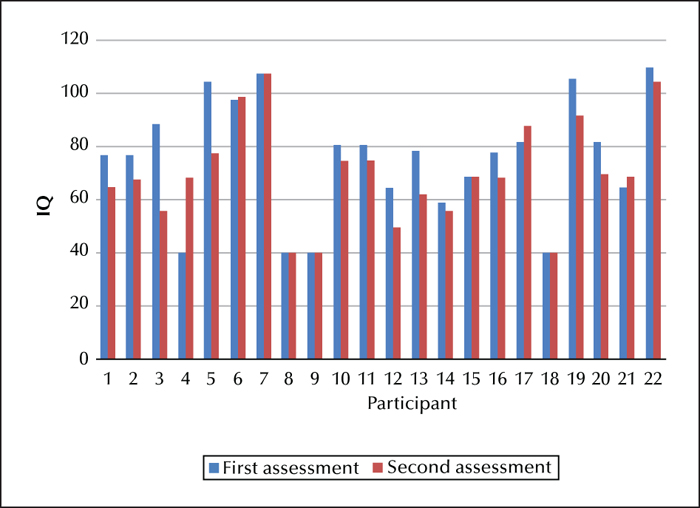
Figure 3 shows the change in IQ for each participant which can be correlated with the clinical histories shown in table 1 (the participants 1-22 in table 1 correspond to those in figure 3).
To address the important issue of statistical versus clinical significance regarding change in IQ scores, we analyzed the data using a conservative criterion of a =>12-point difference in IQ. When adopting this more conservative criterion to denote clinically significant change, seven participants were found to show a =>12-IQ point change with a mean FSIQ at initial assessment of 86 (sd=15), and at follow-up of 67 (sd=14.7) with a large effect size (d=1.3). The largest effect was observed in Performance IQ with a mean reduction of 24 points from 92 to 68 (p=0.018; d=1.5). A less dramatic drop in VIQ was observed with an average of a 9 point-change (d=0.4). If excluding those participants who scored at floor value at both time points (n=4), our findings suggest that 7/18 participants (38%) showed a highly significant intellectual decline affecting FSIQ and PIQ. These individuals tended to move from the average range to the mild learning disability range. Four individuals had IQ values =
Discussion
We report the largest neuropsychological outcome study of childhood ESES with main findings of a clinically and statistically significant decrease in Full-Scale and Performance IQ despite treatment. Around one-third (7/22) of participants in the current study showed a highly significant loss (=>12pts) in intellectual performance, evident particularly within their Performance IQ scores within a relatively short period of time (2.5 years). These individuals tended to move from the average range to the mild learning disability range and were not distinguishable from the rest of the sample in terms of clinical history, imaging or duration of ESES. Performance IQ (PIQ) effects were more pronounced than for verbal IQ, a finding of interest as language difficulties are the most commonly reported difficulties in these children, and may reflect reduced processing speed, working memory and overall cognitive efficiency.
Whilst there have been several descriptions of the clinical and behavioural manifestations of ESES, few have used standardized tests to look at cognitive function. In a report of longitudinal neuropsychological outcome in 10 patients followed from the time of diagnosis into adulthood (mean follow-up duration of 15.6 years), six patients had an IQ <70, two had a low IQ (77 and 84), and two had an IQ within the average range (103 and 85) (Seegmuller et al., 2012). The authors also describe social, cognitive and attention difficulties in the majority of the patients. They suggest that the initial severity and duration of the acute phase regression and the duration of ESES were most predictive of poorer neuropsychological outcome. In another report it was suggested that the initial diagnosis had little prognostic significance, but that length and the age at onset of CSWS, the site of epileptiform activity and the individual neuropsychological profile are more useful for predictors of long-term cognitive outcome (Veggiotti, 2012).
Our study has limitations as a result of the retrospective nature and lack of control over timing of assessments and therapeutic interventions. We relied on a single outcome measure, i.e. IQ testing, and whilst the predictive utility, validity and reliability of standardized IQ tests are high (Dorris et al., 2017), it would be useful for future studies to include measures of adaptive behavior, Quality of Life and psychiatric diagnoses via standardized instruments in order to develop a comprehensive model of impact for affected children and families. The value of the current report lies in the serial neuropsychological evaluations in individuals which demonstrate the clinical impact of ESES. Controlled trials in ESES are required to help determine what might be the most effective treatments. Recently, the RESCUE ESES international European collaboration has developed such a trial. Unfortunately, due to the rarity of the condition, the challenges of delivering multi-centre international trials (see Gentry et al., 2018), and variability in resources to support the trial assessments, this study has not recruited as well as planned. It is possible that international agreements between collaborating centres to collect data and manage patients in a limited number of standardized paradigms might be the most practical way to generate sufficient clinically useful data on treatment efficacy.
The implications of our study are that whilst some children experience stagnation in development, represented by a plateau in neuropsychological test scores and a corresponding decline in age-normative standard scores, others experience a more significant decline in cognitive functions consistent with an active disease process. In this group of children with ESES with a variety of underlying aetiologies and treatments, the underlying encephalopathy resulted in an insidious decline in cognitive ability. It is therefore important that children with ESES have early and continuing neuropsychological assessment and that developmental monitoring is linked with appropriate psychological, social and psychiatric support to improve mental wellbeing and social participation. Families and educational services should also be provided with accurate information to inform prognosis and support in relation to maximizing learning potential and social integration with peers.
Disclosures
None of the authors have any conflict of interest to declare.