Epileptic Disorders
MENUPhenotypic spectrum and long-term outcome of children with genetic early-infantile-onset developmental and epileptic encephalopathy Volume 24, issue 2, April 2022

Epilepsy is a brain disease that can be defined by either at least two unprovoked (or reflex) seizures occurring >24 hours apart, or one unprovoked (or reflex) seizure with a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years, or diagnosis of an epilepsy syndrome [1]. A certain cluster of early-infantile-onset epilepsy syndromes has been grouped as “developmental and epileptic encephalopathies (DEEs)” with highly heterogeneous clinical features, including early myoclonic encephalopathy (EME), Ohtahara syndrome (OS), West syndrome (WS), Dravet syndrome (DS), malignant migrating focal seizures of infancy (MMFSI) as well as non-syndromic DEEs [2]. The early-infantile-onset DEEs are one of the most devastating early-onset epilepsies that contribute to a progressive decline of cerebral function. These are often characterized by a peak of seizures at ∼six months with refractory seizures, severe electroencephalographic (EEG) abnormalities, and developmental delay or intellectual disability [3]. The underlying aetiology of early-infantile-onset DEEs is complex and diverse, ranging from infectious factors, immune disorders, structural abnormalities, metabolic disability, genetic defects and unknown factors. In particular, extensive interest has been devoted to genetic aetiology and many gene mutations have been identified. It turns out that at least 20-30% of early-infantile-onset DEEs are caused by a single gene variant [4]. In recent years, an increasing number of novel genes has been identified in early-infantile-onset DEEs. Many gene variants related to early-onset DEEs have been detected, such as those of SCNIA, SCN2A, SCN8A, STXBP1, CDKL5 and KCNQ2 [5, 6]. Nevertheless, many mutations in patients with genetic early-infantile-onset DEEs are sporadic, occurring in patients with no family history of seizures or epilepsy. Although genetic early-infantile-onset DEEs are increasingly being identified, there is considerable genetic heterogeneity as well as phenotypic heterogeneity. For instance, several cases of dyskinesia, which is a relatively rare clinical symptom, have been identified in early-infantile-onset DEEs [7], and other specific phenotypes have been associated with genetic early-infantile-onset DEE with burst suppression (DEE-BS) [8, 9]. However, the long-term outcome of genetic early-infantile-onset DEEs remains unknown. Hence, it is necessary to gain insight into the broader clinical spectrum, specific genotype-phenotype and long-term outcome of genetic early-infantile-onset DEEs. In this study, we aimed to describe the clinical features and long-term outcome of a cohort of patients with genetic early-infantile-onset DEEs, followed for a period of up to 10 years.
Materials and methods
Patients
This retrospective study included children with genetic early-infantile-onset DEEs at the Department of Neurology, Children's Hospital of Fudan University. The project was approved by the Ethic Committees of Children's Hospital of Fudan University. All experimental protocols involving human subjects were in accordance with guidelines of the institutional and/or national research committee and with the Declaration of Helsinki. Informed consent was obtained from all subjects.
The inclusion criteria for patients were as follows:
- •seizures within six months after birth;
- •frequent seizures; weekly or daily pharmaco-resistant seizures;
- •developmental delay, stagnation or regression;
- •as well as EEG showing frequent epileptic discharges (confirmed by multiple and multifocal sharp waves, spikes, and sharp or spike slow waves).
- •The exclusion criteria for patients were as follows:
- •perinatal brain injury;
- •metabolic disease;
- •intrauterine infection;
- •as well as neonatal and infantile seizures caused by brain structural abnormalities or cortical dysplasia.
This retrospective hospital-based single-centre observational study included 470 patients with early-infantile-onset DEE for genetic testing, according to the above inclusion and exclusion criteria. The clinical data of 470 affected patients between January 2010 and January 2020 were obtained. The age of patients ranged from 3 to 16 years old.We focused on phenotypic parameters, including birth history, seizure onset age, seizure semiology, dyskinesia, EEG, brain magnetic resonance imaging (MRI) findings, clinical genetic evaluations as well as specific syndromes. All MRI scans were assessed based on overall volume, ventricle size, sulcal widening, callosal malformation, cortical morphology, myelination, and basal ganglia, at least one to three times. EEG was used for syndrome classification and assessing treatment response. Scores for the Gesell Developmental Scale or Wechsler Intelligence Scale were used to estimate the development of intelligence in patients with early-infantile-onset DEEs. Outcomes, including seizure frequency, response to therapy and mortality, were also considered. Seizure control was defined for patients without a seizure for a period three times longer than the maximum inter-seizure interval before treatment, or without a seizure for more than one year. All patients were followed via telephone and outpatient or inpatient departmental visits at least once every 6 to 12 months. All patients were followed for one to 10 years.
Next-generation sequencing and copy number variation
The peripheral blood samples of 470 children and their parents were collected. Trio-based whole-exome sequencing and analysis of copy number variation were subsequently performed on all subjects and their parents to screen for pathogenic mutations. According to the criteria for classification of genetic variation and guidelines of the American College of Medical Genetics and Genomics (ACMG), the pathogenicity of variants was identified and evaluated. Effect on protein function caused by gene variants was predicted using SIFT, PolyPhen2, LRT, MutationTaster, FATHMM and other protein function prediction software. Alterations were excluded based on the following criteria: (1) copy number variations, microdeletions or microduplications; (2) nucleotide variation in all normal controls; (3) synonymous mutations; as well as (4) single-nucleotide polymorphisms (SNPs) annotated in the human gene mutation database (HGMD), thousand human genome database, PubMed database and UCSC database. We validated parental origin of variants using PCR-Sanger sequencing and identified putative causal mutations according to parental origin of the variants and clinical features of all patients.
Results
Patient demographics and clinical features
Among 470 patients, 128 showed gene mutations (128/470; 27.2%). Of these 128 patients, 10 were found to have copy number variations. The remaining 118 patients diagnosed with genetic early-infantile-onset DEEs were therefore analysed. Among the 118 patients, 62 (62/118; 52.5%) were male and 56 (56/118; 47.5%) were female. The mean (±SD) age at seizure onset was 3.5 (±1.5) months (range: one day to six months). None of the parents were closely related to one another, and the children were unrelated except for Patients 73 and 74. All patients were followed for one to 10 years. The mean follow-up period was 7.7 years.
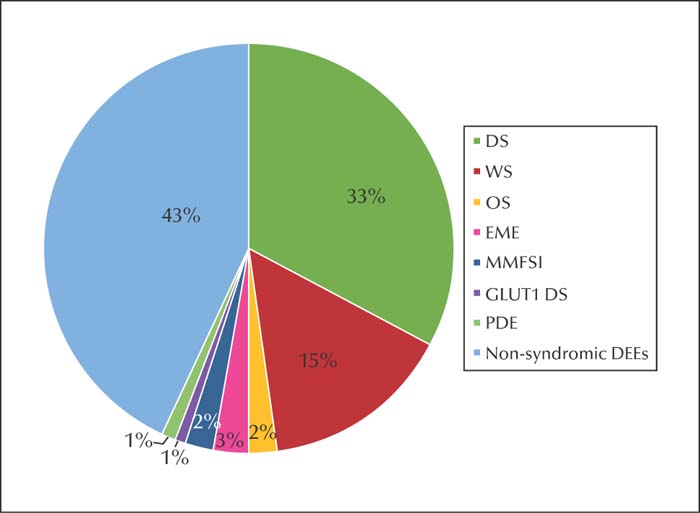
Thirty-nine patients (39/118; 33.1%) were diagnosed with DS, 18 (18/118; 15.3%) with WS, three (3/118; 2.5%) with OS, three (3/118; 2.5%) with EME, two (2/118; 1.7%) with MMFSI, one (1/118; 0.8%) with glucose transporter type 1 deficiency syndrome (GLUT1 DS), one (1/118; 0.8%) with pyridoxine-dependent epilepsy (PDE), and 51 (51/118; 43.3%) with non-syndromic early-infantile-onset DEE (figure 1). Infrequent epileptic discharges on EEG were observed in 74 patients, whereas 18 patients showed normal EEG. A total of 112 patients (112/118; 94.9%) showed normal brain MRI, and the remaining six had widened extracerebral space. Later on, 115 patients were re-examined by brain MRI, one to three times, revealing the widened gap to be normal, and only two patients had mild brain atrophy. After antiepileptic treatment, seizures were controlled in 42 patients (42/118; 35.6%). Importantly, seizures were controlled in 16 patients (16/118; 13.6%) for more than one year, in three patients (3/118; 2.5%) for more than two years, in nine patients (9/118; 7.6%) for more than three years, in eight patients (8/118; 6.8%) for more than four years, in four patients (4/118; 3.4%) for more than five years, and in two patients (2/118; 1.7%) for more than six years. Two (2/118; 1.7%) patients died from status epilepticus. At the final follow-up visit, these patients remained seizure-free with no remarkable improvement in development of intelligence. Among 39 patients with DS, seizures were controlled in five. For 38 patients diagnosed with DS and SCN1A variants, the datais not listed in supplementary table 1 because their clinical features were easily identified. The features of the remaining 80 patients are summarized in supplementary table 1.
Genetic analysis
SCN1A mutations were detected in 38 patients (38/118, 32.2%), representing the largest proportion, including 27 missense, seven frameshift and four nonsense mutations. Genetic causes of DEEs were shown to involve pathogenic mutations (55 missense, 11 frameshift, 11 nonsense, and three splicing mutations). The genetic findings of all 80 patients are summarized in supplementary table 2. The identified genes are summarized in table 1; the most frequently mutated gene in our study was SCN1A (38/118; 32.2%), followed by KCNQ2 mutations in nine patients and CDKL5 mutations in eight patients. Genes associated with ion channels accounted for the highest occurrence (66/118; 55.9%). A summary of the mutated genes is presented in in table 2.
Early-infantile-onset DEE-BS
After performing genetic tests, six patients were found to have KCNQ2 mutations and the remaining mutations were identified in SCN2A (n=2) and STXBP1 (n=1). In our study, children with early-infantile-onset DEE-BS exhibited various clinical phenotypes. The common phenotypes were OS and non-syndromic early-infantile-onset DEE. A total of seven patients with early-infantile-onset DEE-BS had persistent BS, lasting for one day to one month, and in one patient this was accompanied by intermittent diffuse low voltage in both hemispheres, and the persistent BS disappeared at two to three months. For the other two patients with SCN2A mutations, BS on EEG was temporarily suppressed during sleep at four months and five months, respectively, and disappeared at seven months. After treatment, seizure control was achieved in only two patients, seizures were uncontrolled in six, and one died from status epilepticus at six months.
Early-infantile-onset DEEs with dyskinesia
A total of seven children diagnosed with early-infantile-onset DEEs were found to have dyskinesia, and the age at onset of dyskinesia ranged from one month to one year. Two patients with dyskinesia showed dancing-like movements, four patients displayed dystonia, and one patient showed ataxia. Genetic mutations were identified in CDKL5 (n=2), SLC2A1 (n=1), STXBP1 (n=2), TBC1D24 (n=1) and GRIA4 (n=1). The dyskinesia patients with CDKL5 encephalopathy were administrated madopar, yet they constantly showed chorea. Administration of baclofen was observed to have good therapeutic effects in four patients with dystonia. Of these four patients, one with STXBP1 encephalopathy showed a significant response to the administration of levetiracetam (LEV).
Early-infantile-onset DEEs limited to females with cluster seizures associated with SMC1A gene mutation
Three patients with heterozygous de novo mutations in the SMC1A gene were reviewed. All patients were females with moderate-to-severe developmental impairment. None of them were diagnosed with Cornelia de Lange syndrome. All three patients had prominent clinical features of cluster seizures. All nonsense mutations were predicted to negatively affect the SMC1A protein. All patients were treated with multiple antiseizure medications (ASMs) but their seizures remained refractory. However, when initiated with the ketogenic diet (KD), the patients became seizure-free within three to four weeks.
Early-infantile-onset DEEs starting with a febrile seizure
The typical clinical feature of DS is the onset of a febrile seizure, often observed at one year of age, which is characterized by repeated generalized or hemiclonic seizures. In addition to DS, we identified another type of early-infantile-onset DEEs with febrile seizure at onset associated with HNRNPU mutation. The index patient was a six-year-old boy, without a family history of DEEs. Developmental milestones showed moderate developmental delay. He first had febrile seizures at six months of age, which occurred five times a day. Video-EEG showed slow background activity and sharp slow waves in the left occipital and posterior temporal regions during the interictal period. A febrile seizure occurred every six months, on average. At the age of four years, he began to have seizures without any inducing factors. EEG showed a large number of multifocal sharp waves, spike waves, and spike-slow waves. Treatment with phenobarbital (PB) and LEV resulted in poor therapeutic efficacy, whereas the frequency of epileptic seizures was reduced with valproate acid (VPA). At the final follow-up visit, he had remained seizure-free for two years with LEV and VPA treatment but there was no remarkable improvement in his development of intelligence.
Successful treatment of genetic early-infantile-onset DEEs
In general, the effect of KD therapy is promising as treatment for genetic early-infantile-onset DEEs. A total of three patients with SMC1A mutations responded well to KD add-on therapy. VPA add-on treatment had a positive effect on KCNB1 (n=2)and PACS2 (n=3) encephalopathy. Moreover, treatment with VPA was also effective for a patient with mutation in HNRNPU (n=1). LEV add-on treatment was particularly effective for STXBP1 (n=2) encephalopathy and oxcarbazepine (OXC) add-on treatment was also of benefit for SCN8A (n=2) encephalopathy.
Discussion
We report a series of individuals with genetic early-infantile-onset DEEs, delineating the phenotypic spectrum and long-term outcome. For cases of early-infantile-onset DEE of unknown cause, the rate of gene mutation detected was 27.2% (128/470). In cases of genetic early-infantile-onset DEE, non-syndromic early-infantile-onset DEEs represented the majority (51/118; 43.3%). The initial EEG of most patients showed frequent multiple and multifocal discharges. Upon seizure control, only a minority of patients showed infrequent epileptic discharges or normal EEG. Despite several brain MRI scans, no significant change in MRI was observed at a later stage. In the long-term, the seizure control rate for genetic early-infantile-onset DEEs was 35.6% (42/118). The death rate was 1.7% (2/118). Genetic analysis showed that SCN1A was the most commonly affected gene (38/118; 32.2%), followed by KCNQ2 mutations (9/118) and CDKL5 mutations (8/118). In line with our study, Symonds et al. also reported that SCN1A and KCNQ2 were commonly mutated genes in early-childhood-onset epilepsy [6]. BS is one of the EEG phenomena in early-infantile-onset DEEs that usually occurs during sleep and awakening in patients with OS and the sleep period in patients with EME. There are two different types of BS patterns, namely early BS and late BS [8, 10]. As for the definition of early BS and late BS, this is not very clear at present. Yoshitomi et al. claimed that BS should be classified according to one month of age [10]. In the present study, we found that BS is not only present in OS, but also in other non-syndromic early-infantile-onset DEEs. Our study provides an in-depth understanding of the genetic factors of early-infantile-onset DEE-BS, and underlines the important role of genetic factors in addition to common causes, such as cortical malformations. In our study, DEE-BS pathogenic mutations were identified in nine patients, accounting for 7.6% (9/118). Moreover, we showed that the largest genetic subgroup of early-infantile-onset DEE-BS was the subgroup with KCNQ2 mutations, accounting for 66.7% (6/9). A total of nine patients with KCNQ2 mutations in this group had various types of seizures with poor outcome and prognosis related to treatment. For cases with early onset of persistent tonic and spasm seizures and other types of intractable seizures, seizures with early BS on EEG may suggest the presence of KCNQ2 pathogenic mutations. However, DEE-BS is highly heterogeneous in terms of genetic aetiology. Following the KCNQ2 variant subgroup, the second largest subgroup was the SCN2A variant subgroup. For two patients with KCNQ2 and SCN2A variants in this study, EEG was suppressed during sleep at four months and five months, and disappeared at seven months. The reason for transient BS in the SCN2A variant subgroup might be attributed to the immaturity of the central nervous system or gene mutation leading to brain dysfunction at this stage. In the third subgroup, a STXBP1 variant was reported in one patient. Early-infantile-onset DEE related to STXBP1 gene mutation has been previously reported, and the common phenotype is OS. Mutations in the STXBP1 gene can cause abnormal neurotransmitter release and brain stem cell apoptosis and dysfunction. This also changes the excitability of neurons resulting in seizures [11]. We also found that early-infantile-onset DEE-BS responded poorly to ASMs. In this study, seven cases of early-infantile-onset DEEs were found to have dyskinesia including dystonia, chorea, paroxysmal dyskinesia, Parkinson's syndrome, ataxia, tremor, etc. The onset age for dyskinesia ranged from one month to one year. A total of three patients were diagnosed with an epilepsy syndrome, namely WS, GLUT1 deficiency syndrome and EME. The main clinical symptoms of dyskinesia were dystonia, chorea and ataxia. Genetic mutations were identified in CDKL5, SLC2A1, STXBP1, TBC1D24 and GRIA4. Our findings indicate that administration of baclofen showed therapeutic effects in four patients with dystonia. One of the patients with STXBP1 encephalopathy and dystonia showed a good response to the administration of LEV. The symptoms of dyskinesia in a few patients were also relieved with the control of epileptic seizures.
SMC1A mutations are only associated with early-infantile-onset epilepsy in female patients with cluster seizures. So far, a spectrum of SMC1A gene variants has been shown to be associated with Cornelia de Lange syndrome (CdLS), epileptic encephalopathy with seizures in female patients, colorectal carcinomas, bladder cancer and leukaemia [12-19]. All our three patients became seizure-free when the KD was used as add-on therapy. SNC1A mutations have been previously reported to be related to oxidative stress in CdLS cell lines [20]. KD treatment in children with refractory epilepsy has also been demonstrated to improve mitochondrial function and decrease oxidative stress [21, 22]. Our results support this regarding the effectiveness of KD therapy.
HNRNPU,located at Chromosome 1, encodes heteronuclear ribonucleoprotein U. It is expressed in the adult brain, heart, kidney and liver, especially in the cerebellum. It was first reported to be related to 1q43-q44 deletion syndrome [23]. Later, a variety of clinical phenotypes related to HNRNPU mutation were reported, mainly including early-infantile-onset epilepsy with severe intellectual disability, WS, DEE, Lennox-Gastaut syndrome and craniofacial deformity [24-26]. Durkin et al. described HNRNPU gene mutation-related disease to more likely be a kind of neurodevelopmental syndrome [26]. The authors reported 21 cases of children, of which three showed onset with febrile seizures. Together with a case in this study, we believe that onset of early-infantile-onset DEE with febrile seizure is a specific phenotype of HNRNPU-related neurodevelopmental syndrome, which is similar to DS.
These genes and their functions have mainly been categorized as genes that relate to ion channels, the synapsis, neurotransmitters and receptors, signal transduction, DNA and RNA regulation, organelles and the cell membrane, and the development and growth of neurons [27]. In this study, we found that ion channel gene mutations accounted for the largest proportion (66/118; 55.9%). Among them, sodium channel gene mutations represented the majority (47/66; 71.2%). In WS, we detectedgene mutations in the following genes: SCN3A, SCN2A, SCN8A, CACNA1H, DEPDC5, MECP2, DYNC1H1, CDKL5, ALG11, CCDC88C, GABAA1, IL1RAPL1, RNASEH2B, SLC19A3, STXBP1, RARS2, and COL4A2. In addition to common gene mutations, we reported rare possible pathogenic gene variants, namely in CCDC88C, IL1RAPL1, RNASEH2B and COL4A2, in patients with WS. For non-syndromic genetic DEEs, we detected rare possible pathogenic gene variants in SETBP1, DPYD, CSNK2B and H3F3A. With regards to inheritance pattern, de novo heterozygous mutations accounted for the largest percentage; 88.1% (104/118). Among these types of mutations, missense mutations constituted the majority. As expected, some of the genes were included in more than one classification group due to multiple functions.
Generally, genetic early-infantile-onset DEEs respond poorly to ASM treatment. However, we found that specific ASMs showed good therapeutic effects against some early-infantile-onset DEEs with genetic variants. As well as the promising effect of the KD as treatment for SMC1A encephalopathy, we also found that VPA add-on treatment showed a positive effect in patients with KCNB1 and PACS2 encephalopathy, and LEV and OXC add-on treatment was effective for STXBP1 encephalopathy and SCN8A encephalopathy, respectively. VPA is a broad-spectrum ASM, which exerts its anticonvulsant effect through a variety of mechanisms. In particular, VPA can regulate the expression of endoplasmic reticulum stress proteins (the 78-kDa glucose-regulated protein [GRP78], the 94-kDa glucose-regulated protein [GRP94] and calreticulin) [28-30]. Moreover, VPA can also regulate the level of intracellular Ca2+ by increasing the expression of endoplasmic reticulum stress proteins, improving the calcium-binding ability of the endoplasmic reticulum, and ultimately enabling cells to adapt to cellular stress caused by imbalance of intracellular Ca2+ homeostasis [30]. The gene, PACS2, is necessary for effective Ca2+ transfer between the endoplasmic reticulum and mitochondria, whereas GRP78 is involved in Ca2+ transport from the endoplasmic reticulum to mitochondria [31-33]. Therefore, we speculate that VPA may suppress PACS2-related DEEs by regulating PACS2-associated cellular functions. In addition, syndrome-specific treatment was also effective against DEEs. For example, the use of stiripentol in combination with VPA and clobazam was effective for DS, and treatment with adrenocorticotropic hormone (ACTH) or vigabatrin alleviated symptoms of WS. However, the underlying mechanism still needs further investigation. Understanding the pathophysiology of the underlying gene defect will certainly help to pave the way towards possible future individualized treatments. Further research should include a larger cohort in order to validate our observations. The mechanism that links rare gene mutations to seizure outcome should be explored in greater detail in future studies.
Conclusion
We describe the clinical features and long-term outcome of genetic early-infantile-onset DEEs. The clinical manifestations of early-infantile-onset DEEs were highly diverse and heterogeneous, as exemplified by the presence of dyskinesia. After appropriate treatment, some patients achieved seizure freedom, nevertheless there was no remarkable improvement in their development of intelligence. The early-infantile-onset DEEs with febrile seizure at onset may represent a specific phenotype of HNRNPU-related neurodevelopmental syndrome, similar to DS. Ion channel gene mutations were most frequently documented. Four rare possible pathogenic gene variants were reported in WS and four in non-syndromic genetic early-infantile-onset DEEs. Although genetic early-infantile-onset DEEs responded poorly to ASM treatment, we found that specific ASMs showed good therapeutic effects against some early-infantile-onset DEEs with genetic variants.
Supplementary material.
Supplementary data and summary didactic slides accompanying the manuscript are available at www.epilepticdisorders.com.
Ethics approval and consent to participate.
The project was approved by the Ethic Committees of Children's Hospital of Fudan University. All the experimental protocols involving human subjects were in accordance with guidelines of the institutional and/or national research committee and with the Declaration of Helsinki.
Acknowledgements and disclosures
We thank all the patients and their guardians who participated in this study. Moreover, our heartfelt gratitude goes to all the team members for their dedicated work.
None of the authors have any conflicts of interest to disclose with regards to this manuscript.
Funding
The project was supported by Shanghai Municipal Science and Technology Major Project (Grant No. 2017SHZDZX01).Key points