Epileptic Disorders
MENUIdentifying patients with epilepsy at high risk of cardiac death: signs, risk factors and initial management of high risk of cardiac death Volume 23, issue 1, February 2021

Learning objectives [1]
- •Acquire competency to distinguish syncope from epileptic seizures.
- •Understand and explain the manifold cardiac complications and their causes in people with epilepsy (PWE).
- •Acquire competency to recognize and anticipate cardiac complications in PWE by thorough history taking, analysing clinical signs of seizures and making use of basic tools such as 12-lead ECG and simultaneous one-lead ECG during EEG recordings.
- •Understand and recognize the potential contribution of antiseizure treatments to cardiac complications in PWE.
- •Acquire competency to take actions to manage and to reduce the risk of cardiac complications in PWE.
People with epilepsy (PWE) have a 2-3-fold increased risk of dying prematurely as compared to the general population, which is, in a significant proportion (15%), due to sudden cardiac death (SCD) or acute myocardial infarctions [2-6]. This may be partly explained by higher prevalence of comorbid cardiac conditions and a higher burden of cardiovascular risk factors [7, 8]. For instance, PWE are more often obese, physically inactive and current smokers, and their cardiovascular risk profile (i.e. hypercholesterolaemia, diabetes mellitus, metabolic syndrome) is worse than in the general population [9-12]. Intriguingly, reports of cardiac arrhythmias and cardiac pathology at post-mortem evaluation in PWE suggest a cardiac component to the pathology of sudden unexpected death in epilepsy (SUDEP) or that at least in some cases, SCD and SUDEP are overlapping entities [13], underscoring the clinical importance to understand the signs and symptoms of cardiac complications in PWE. In addition, the increased mortality of status epilepticus may also be linked to cardiovascular complications, as recently reviewed [14]. The causes of elevated cardiovascular morbidity and mortality in PWE are manifold and include detrimental effects of epileptic seizures, treatment and epilepsy itself [15-19]. Given the potentially severe consequences of cardiac conditions, we summarize in this educational review the many facets of cardiac complications and their underlying causes, and explain how to recognize and manage them to mitigate the cardiac risks in PWE.
The spectrum of cardiac complications in epilepsy
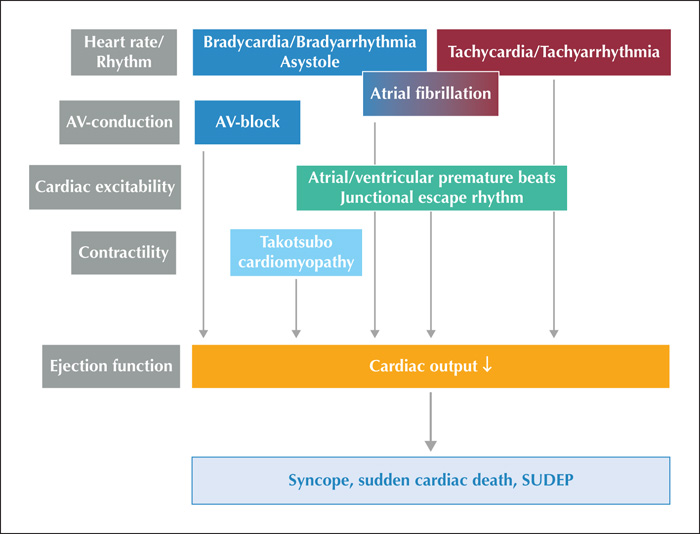
PWE can of course suffer from all types of cardiac pathology including infection and inflammation of the pericardium, myocardium and endocardium, abnormalities of the cardiac valves, coronary artery disease (CAD), and cardiac arrhythmias. The risk of the two latter, however, is significantly elevated in PWE [16, 19], prompting the need for systematic assessment of putative causes and related signs in PWE.
CAD is due to obstructions of the coronary arteries, leading to insufficient blood flow and impaired oxygen supply, which in turn causes reversible or irreversible myocardial ischaemia. Classic signs of symptomatic CAD include chest pain, elevations of circulating cardiac injury markers such as troponin, and typical ECG alterations such as ST segment elevation. Seizure-related sustained, generalized muscle activity along with profound cardiorespiratory dysfunction bear, in the presence of CAD, an elevated risk of myocardial ischaemia (see section on Seizure-related cardiac complications).
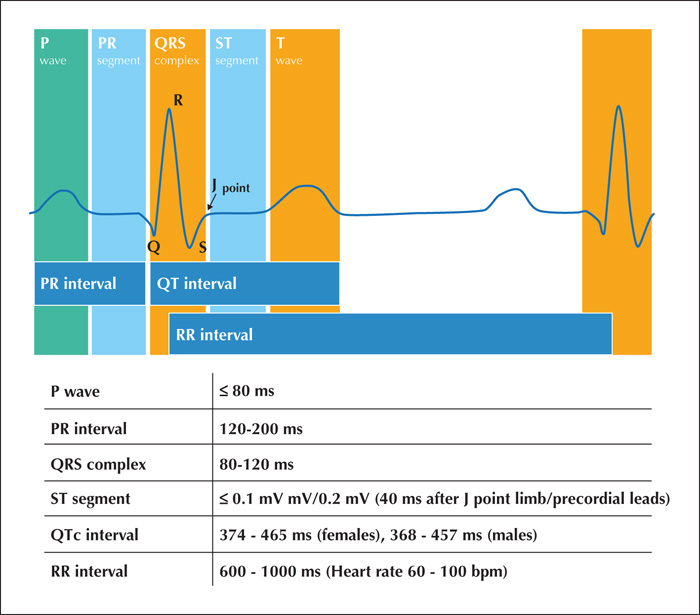
Cardiac arrhythmias are due to altered electrical properties of cardiac tissue leading to disturbed excitation and conduction in the atria and ventricles; heartbeats can be slowed (bradyarrhythmia, asystole) or accelerated (tachycardia), regular or irregular, and the coordinated contractions of atria and ventricles can be disturbed (table 1). Typical clinical manifestations include palpitations, symptoms and signs due to syncope (see below). Cardiac arrhythmias may also manifest indirectly by embolic events (e.g. thromboembolic stroke due to atrial fibrillation). ECG recordings are key to the diagnosis of cardiac arrhythmias, and the interpretation of a 12-lead ECG is part of the general skills of physicians (figure 1, table 2). Most, if not all forms of cardiac arrhythmias were described in PWE in the absence of, or in association with seizures (figure 2) [17, 20]. If the extent and duration of these cardiac alterations are large and long enough, they lead to a significant reduction of cardiac output with a subsequent drop in systemic arterial blood pressure (BP). This, in turn, causes hypoperfusion of body organs including the brain, heart and kidneys (figure 2). If the hypoperfusion is sufficiently long or sustained, this leads to irreversible damage of all end organs and to death. In contrast, a transitory drop of global cerebral perfusion is linked to a transient loss of consciousness (TLOC) only, which is called syncope (see below). As symptoms of syncope resemble those of epileptic seizures, it can be challenging to make the correct diagnosis of syncope especially in PWE. At the same time, syncope of different origins can be mistaken for epileptic seizures, leading to the false diagnosis of epilepsy. Maybe the most difficult challenge is to make the correct diagnosis of ictal syncope, i.e. syncope that is induced by an epileptic seizure (see below), which is typically made in the setting of continuous video-EEG monitoring (VEM).
Syncope
Syncope results from global cerebral hypoperfusion which, in turn, is caused by a temporary low systemic arterial BP. The decrease in BP may provoke a range of symptoms and signs, dependent on the severity and the speed of the BP drop and the ability of the cerebral autoregulation to compensate for the low BP [21]. The symptoms and signs of syncope can be categorized into two groups. The first group comprises the consequences of the cerebral or retinal hypoperfusion due to the failure of the systemic perfusion. These features are similar for all causes of syncope. The second group are the complaints specific to the form of syncope and will be detailed later.
Patients will only report symptoms of cerebral or retinal hypoperfusion if the fall in arterial pressure is slow enough to perceive symptoms and to “store” the sensations for later recall [22]. Conversely, if the fall in arterial BP is too steep, syncope may occur without prodromes. Subjects may even be unable to recall loss of consciousness (LOC) (‘amnesia for amnesia’) and thus present with unexplained falls. This presentation is more common in the elderly. As BP decreases, subjects often report ’light-headedness’ or dizziness as the first symptom of low cerebral perfusion. Other descriptions include ’fading out’ or ‘emptiness in the head’. If BP drop is slow, blurred vision and loss of peripheral vision may ensue as a second symptom relating to retinal hypoperfusion. Some subjects report “grey out” (loss of colour vision) or “black out” (darkened vision). Hearing loss may occur following loss of vision. With further BP decline, subjects have difficulty concentrating and become unaware of their surroundings. A brief period of staring and an inability to act may occur prior to complete syncope [22]. Eventually, when BP reaches the critical closing pressure, patients will fall and lose consciousness. LOC usually ensues when systolic BP is 60 mm Hg or lower but may also occur at higher BP when the BP drop is steep [23].
The clinical signs are linked to the severity of the cerebral hypoperfusion and reflected by the EEG patterns seen in syncope. Mild cerebral hypoperfusion will cause a generalized “slow” EEG pattern (a short-lasting period of abnormal delta waves), whereas in severe hypoperfusion, a “slow-flat-slow” pattern is seen; the generalized slow pattern is followed by a generalized flattening of the EEG, in turn, followed by a slow pattern of delta waves as the cerebral perfusion restores and then return to a normal EEG pattern. The EEG flattening is mostly seen in syncope involving asystole (e.g. cardioinhibitory vasovagal syncope), but can also occur in vasovagal syncope without asystole, other arrhythmias and in orthostatic hypotension [23]. The EEG only reflects the cortical brain functions. Signs during a flat EEG can thus not be explained by cortical functions and likely point towards brain stem activity. Signs of brainstem dysfunction appear later than signs of cortical dysfunction, probably because the brainstem is more resistant to ischaemia. Some clinical signs are mostly seen during the slow phase of the EEG, some mostly during the flattening of EEG and some clinical features are present during both phases [21].
LOC usually starts in the slow phase. Eye opening and loss of muscle tone (e.g. head dropping) and a vacant expression often constitute the very first signs and are usually followed by many other motor phenomena, varying between subjects and between events [22]. Myoclonic jerks are seen in about half of the subjects and usually appear after the subject has fallen [24]. The jerks are strongly associated with the slow phase of the EEG. If a second bout of myoclonic jerks occurs, this starts during the second slow phase. The jerks rarely occur during the flat EEG, thus indicating that their presence signifies decreased but not complete shut-down of cortical activity. The number of jerks is rarely more than 10 [24]. The jerks are typically asynchronous and can be restricted to one side. The eyes are often closed before LOC, but open at onset of unconsciousness, with the pupils mostly directed upwards. If head turning is present, it starts quite early relative to the slow phase. Oral automatisms are seen in about half of the subjects and may occur both during the slow phase of the EEG and during the flat phase [21]. Snoring, gasping, roving eye movements, making sounds and posturing are strongly associated with EEG flattening and thus probably reflect brainstem activity. The duration of unconsciousness rarely exceeds one minute. Eyewitnesses may, however, report longer durations due to the emotional impact of the situation [25]. Subjects rarely bite their tongue; if tongue biting occurs, it is the consequence of the fall and concerns the tip of the tongue. Urine incontinence may occur during syncope, but faecal incontinence is rare [26].
In all forms of syncope, patients recover quickly and are almost immediately clear-headed. If any disorientation occurs, it would likely last [27], or when an abrupt fall will be responsible for a significant head trauma. The duration of syncope is usually too short to cause observable brain damage. It has, however, been demonstrated that the risk of white matter lesions is slightly increased in those with frequent (more than five events of) syncope [28].
Reflex or vasovagal syncope (VVS)
The signs and symptoms of VVS can be divided into two groups. The first concerns the symptoms and signs due to retinal and cerebral hypoperfusion, as described before. The second group is due to the underlying cause: an abnormal reflex of the otherwise normal autonomic nervous system. This ‘vasovagal’ reflex is characterized by vagally (parasympathetically) mediated bradycardia or even asystole with or without withdrawal of the sympathetically driven vasoconstriction. These changes in autonomic activity cause a specific set of signs and symptoms, labelled as ’autonomic activation’, including perspiration, facial pallor, nausea, vomiting, and pupillary dilatation. Autonomic activation is often reported by younger subjects with VVS but less often by the elderly, due to the inability to recall or identify these symptoms or due to mere absence of these signs in the elderly.
It is essential to always enquire about the circumstances in patients with TLOC as VVS never occurs randomly [26]. It is usually precipitated by emotional triggers (e.g. fear, pain, emotional distress or their anticipation) or triggers that facilitate venous pooling (e.g. upon standing, prolonged standing, heat, cessation of exercise, coughing, defecation, swallowing, micturition) or stimulate the carotid sinus (pressure on the neck). At times, various triggers may have a cumulative effect (e.g. watching an emotional scene on the television in a sitting position as an orthostatic trigger). Warning symptoms (including autonomic activation) start on average between 30-60 seconds before the actual onset of unconsciousness but may vary between individuals. Sweating, pupillary dilatation and deathly pallor are frequent early signs [21, 23]. Patients may be able to prevent actual syncope if they recognise these symptoms at an early stage and if they act immediately by sitting, squatting or lying down. Counterpressure manoeuvres (e.g. leg crossing) are also effective to prevent further TLOC. In some patients, the BP remains low after the patient has regained consciousness, resulting in a recurring tendency to faint every time he or she is returned to the upright position (’status vasovagalis’) [27]. A noticeable scarlet flush can occur after VVS if the BP recovery is very rapid (cardioinhibitory vasovagal syncope), but is also seen in those with an arrhythmogenic syncope [23]
Case 1
A 25-year-old woman suffered since adolescence from recurrent episodes characterized by an non-specific feeling in the head (“something is wrong, something will happen”) followed by loss of consciousness. During these episodes, she frequently fell without injuries. Witnesses reported motor phenomena such as generalized stiffening or myoclonic jerks, and on one occasion she had enuresis, but never a tongue bite. Upon recovery, she often heard very loud noises, she had a feeling of warmth and was often fearful for several seconds. The episodes mostly (if not always) occurred in the context of fear or pain. The duration of the episodes was estimated between 1-2 minutes with a frequency of 2-3 per year. Brain MRI and routine EEG examinations were normal. Epilepsy of unknown aetiology was suspected and she was put on 150 mg lamotrigine.
The patient was seizure-free for more than a year before admission for further evaluation. A venous puncture was performed during video-EEG monitoring which provoked an habitual episode. She had her typical “cephalic aura” followed by loss of consciousness. Her arms stiffened with some posturing (slowly raised), followed by a flaccid phase without apparent movements, transitioning again into stiffening of the torso and both arms. Upon recovery of consciousness, she presented with a fearful face, but could adequately respond within 20 seconds. The simultaneous one-channel ECG recordings showed a progressive decline of heart rate, followed by an asystole of 21 seconds, before regular QRS complexes occurred again. The EEG displayed typical signs of syncope with generalized slowing and rhythmic delta activity for some seconds, followed by a flat EEG, generalized rhythmic delta activity and again physiological resting activity.
It was concluded that the patient suffered from recurrent vasovagal syncope linked to emotional triggers, and lamotrigine was tapered off.
Orthostatic hypotension
Orthostatic hypotension (OH) may cause syncope but often the core symptom is orthostatic intolerance with recurrent presyncope due to prolonged severe hypotension. As in VVS, it is essential to enquire about triggering factors [26]. All triggers that are relevant in VVS may trigger OH, expect for emotional precipitants and those related to the stimulation of the carotid sinus. Some presyncopal symptoms are unique for OH, such as the “loss of ability to act”. This is a state in which the BP is too low to think clearly but not low enough to cause LOC. Eyewitnesses may notice a vacant expression and an inability to respond. Often, these events go unrecognised, especially in those with comorbid movement disorders, e.g. Parkinson's disease. Another clinical feature specific to neurogenic OH is the ’coat hanger sign’; pain in the neck radiating to the occipital region and shoulders caused by local ischaemia in the upright position. Pain in the lower back, buttocks and chest during orthostatic stress has also been described in OH but the underlying mechanism is unclear. All symptoms resolve when lying down. Transient ischaemic attacks may occur during periods of hypotension, especially if the patient has carotid occlusive disease (hypotensive transient ischaemic attack).
Cardiac syncope
Cardiac syncope can be caused by arrhythmias, structural heart disease or disorders of the great vessels and cardiopulmonary disease (e.g. acute aortic dissection). Cardiac syncope usually causes an abrupt TLOC without any specific precipitating event. Exceptions to the rule include syncope during exertion, startling (e.g. via an alarm clock) or having cold water on the face [26]. TLOC in the supine position should also raise suspicion for a cardiac cause as it makes VVS or OH less likely given the absence of an orthostatic trigger. Due to the abrupt onset of syncope, patients with an arrhythmogenic cause of syncope usually experience no prodromes or a very short prodromal phase. Patients with syncope secondary to structural cardiac, cardiopulmonary and great vessel disease may, however, experience longer prodromal symptoms. These symptoms may relate to cerebral hypoperfusion or hint towards the underlying cause (shortness of breath, chest pain). The duration of the LOC strongly correlates with the duration of the cardiac standstill or loss of cardiac output. Recovery is marked by facial flushing if the systemic circulation is rapidly restored e.g. in those with a transient AV block [21, 23].
Contrasts with epilepsy
Generalized or focal to bilateral tonic-clonic seizures (TCS) could be misdiagnosed as reflex syncope, as motor phenomena also frequently occur in the latter. Nonetheless, the semiology differs between the two, and one important feature is the number of jerks; fewer myoclonic jerks are seen in vasovagal syncope than in epileptic seizures. In this regard, the “10/20 rule” has been proposed [24]. There is a lack of overlap in the number of jerks between VVS and epileptic seizures, with less than 10 jerks pointing towards reflex syncope and more than 20 jerks towards a TCS. The motor phenomena start in syncope after the fall of the patient, whereas in TCS, the jerks may start before the onset of unconsciousness. The presence of urine incontinence does not discriminate between syncope and seizures [26]. A lateral tongue bite, a long period of postictal coma, confusion or amnesia are, however, strong clues favouring TCS [26]. The typical clinical features of syncope and the clues to distinguish syncope from TCS are summarized in table 3.
Seizure-related cardiac complications
Epileptic seizures, focal and generalized, are known to induce autonomic imbalance [29]. Activation and inhibition of the sympathetic and parasympathetic branches of the central autonomic nervous system commonly occur during and after seizures [30], involving the areas that control heart activity including the insula and hippocampus [31-33]. During most seizures, sympathetic outflow increases, which is reflected by tachycardia, tachypnoea, increased blood pressure, facial flushing, pupillary dilatation and diaphoresis [30]. Parasympathetic activation can also predominate, which involves bradycardia, bradypnea, miosis and increased salivation [30].
Ictal tachycardia
Seizure-related cardiac and respiratory changes were extensively studied in the last decade, in particular because of their potential role in SUDEP and seizure detection. Peri-ictal cardiac arrhythmias are common (figure 2) [20]. The commonest arrhythmia is ictal tachycardia. Ictal tachycardia is seen in up to 80% of all seizures and in 82% of PWE [34, 35]. Ictal tachycardia is usually asymptomatic (thus no true complication), and potentially helpful to automatically detect seizures based on heart rate features using wearable devices [36].
4.2 Ictal bradyarrhythmia
Bradycardia and asystole can be caused by silencing sinus node activity in the right atrium or by blocking the atrioventricular (AV) conduction system. Ictal asystole is defined in most studies as heart rate with RR intervals longer than 3 seconds. It was reported to occur in 0.32% of people with difficult-to-treat focal epilepsy admitted for VEM, mostly in people with temporal lobe epilepsy [20]. Suppression of sinus node activity appears to be the predominant cause of ictal asystole, but complete AV block was also described during focal seizures in a few non-SUDEP cases [20]. Ictal asystole can cause falls and injuries due to seizure-induced syncope. For many years, ictal asystole was thought to be a possible mechanism of SUDEP. In all but one of the reported cases, however, the ictal asystole was self-limiting [20]. In this one case, the asystole lasted for 44 seconds, after which the person was resuscitated successfully [37]. The case was classified as ‘near-SUDEP’ [38]. The longest ictal asystole reported lasted 96 seconds and was self-limiting [39]. One case with ictal asystole fell victim to SUDEP despite a well-functioning pacemaker, suggesting ictal asystole was benign and did not cause death [40]. This case suggests that terminal asystole in SUDEP is not primarily a cardiac mechanism.
Ictal asystole with subsequent syncope (‘ictal syncope’) is characterised by sudden loss of muscle tone during a focal seizure with impaired awareness, commonly originating in the temporal lobe [20, 41]. Serious injury may occur, contrary to VVS, as individuals will not be able to experience any of the classic prodromal signs [21]. It is difficult to make the correct diagnosis of ictal asystole, but the onset of new symptoms (e.g. unusual pallor, flaccid falls with injuries) after the habitual aura should prompt further examinations. The recurrence risk of ictal asystole amounts to 40%, underscoring the need for a timely diagnosis [42]. It is estimated that recording of at least three habitual seizures during VEM is required to allow an accurate diagnosis [42]. Because of the frequent falls and injuries, prevention of ictal syncope is important. Retrospective studies suggest that improving seizure control may prevent ictal asystole [43-45]. If the person is refractory to medical treatment and not suitable for epilepsy surgery, implantation of a cardiac pacemaker should be considered to reduce risk of falls and injuries [45-47]. Contrary to the self-limiting nature of ictal asystole, 54% of reported postictal asystole cases died from SUDEP [20]. Postictal bradyarrhythmias are exceedingly rare and always accompanied or preceded by decreased respiratory rate and apnoea [20, 48, 49]. Although postictal bradyarrhythmias seem to play a crucial role in the cascade of events leading to SUDEP, they seem strongly linked to respiratory dysfunction [49, 50].
Case 2
A 56-year-old woman suffered from difficult-to-treat temporal lobe epilepsy of unknown origin for seven years. Seizures were characterized by sudden loss of awareness without aura, followed by oro-alimentary and manual automatisms and postictal disorientation without speech disturbances. Sometimes, atonic falls were observed by family members.
She was referred for pre-surgical assessment. Brain MRI showed a slight enlargement and signal enhancement of the right temporo-mesial structures. Cerebral FDG-PET displayed moderate temporal hypometabolism on both sides, and neuropsychological testing did not reveal localizing clues. Interictal EEG showed sleep-activated sharp waves in the anterior temporal region on both sides. Prolonged video-EEG monitoring was performed to record seizures. On Day 5, a habitual temporal lobe seizure, with sudden onset of staring, oro-alimentary and manual automatisms, occurred. The seizure started with rhythmic delta activity in the right temporal region with rapid involvement of the contralateral temporal lobe. About 25 seconds after seizure onset, the heart rate progressively declined, followed by an asystole of 30 seconds, before regular QRS complexes re-occurred. These cardiac changes were accompanied by EEG signs typically linked to syncope (generalized rhythmic delta activity, flat EEG, rhythmic delta activity). During the syncopal period, she fell flaccidly onto the bed. The next morning, she suffered from another habitual temporal lobe seizure, again with self-limiting ictal asystole, but this time with a consecutive fall to the floor without injury.
It is important to note that recurrent ictal asystole was only recognised following video-EEG monitoring and that she previously reported falls during her habitual seizures. Because of the medical refractoriness and the rather low chance of achieving seizure freedom upon medical treatment, she was offered a cardiac pacemaker. After implantation, she never experienced falls again during focal seizures. Further intracranial presurgical investigations were denied.
Atrial fibrillation
Atrial fibrillation is characterized by an irregular, slow, or rapid beating of the atrial heart chambers. It may only manifest with palpitations but can also induce dyspnoea or syncope due to impaired cardiac output (if heart rate is low or too fast). Importantly, atrial fibrillation is also an important risk factor for thromboembolic ischaemic stroke because of the desynchronised atrial contractions, allowing formation of cardiac thrombi, requiring appropriate anticoagulation [51]. In general, atrial fibrillation was described in up to about 10% of people with epilepsy; seizure-related atrial fibrillation was mostly described after (focal to) bilateral TCS [20, 52, 53]. Long-term administration of beta-blockers (used for rate and rhythm control) should be considered in cases with difficult-to-treat epilepsy and seizure-induced atrial fibrillation [51, 53].
Ictal ventricular tachycardia or fibrillation
Seizure-induced ventricular tachycardia or fibrillation (VT/VF) were described in six cases that led to SUDEP or near-SUDEP [54-59]. In all these cases, VT/VF occurred directly after a TCS. Signs of a prior myocardial infarction were found in only one SUDEP case (chest pain during a seizure cluster leading up to the fatal seizure) [54]. In the remaining five cases, no underlying cardiac pathology was detected, suggesting that seizure-related alterations of electrical properties have facilitated onset of VT/VF. Indeed, profound changes of cardiac repolarisation indices, known as established markers of elevated risk of VT/VF and SCD, occur especially in association with TCS [60, 61]. This is probably linked to the strong sympathetic activation and massive catecholamine release (up to three-fold) during TCS [62]. The catecholamine levels normalise within two hours after the TCS, indicating a transient hyperadrenergic state [62]. Transient abnormal prolongation of QT intervals occurs in about 6% of the seizures in 12% of the patients, and abnormal shortening of the QT interval are commonly observed in the aftermaths of most focal to bilateral TCS [60, 61]. Furthermore, T-wave alternans (TWA) (see section on Epilepsy-related cardiac complications) is significantly increased following TCS, possibly increasing the risk of VT/VF and SCD [63]. Fortunately, seizure-induced VT/VF seems very rare. There could be, however, a publication bias, as fatal VT/VF cases may not be categorised as SUDEP. In a prospective study of (out-of-hospital) cardiac arrests after VT/VF, it was found that the risk of VT/VF was three-fold higher in PWE than the general population [5]. Most of those VT/VF cases occurred in patients with (pre-existing or acute) heart conditions, but some were unexplained and could be classified as near-SUDEP or SUDEP [64]. SCD and SUDEP thus seem to partly overlap.
Acute myocardial injury and infarction
It is plausible to assume that generalized onset and focal to bilateral TCS favour cardiac oxygen deprivation by inducing strong muscle activity, apnoea and prominent, sustained sinus tachycardia [35, 61] In line with this assumption, transitory ECG changes, possibly indicating cardiac ischaemia such as ST segment depression and T wave inversion, were described particularly in association with focal to bilateral TCS, although such ECG signs appear to be rather infrequent [60, 62, 65, 66]. Elevated levels of high-sensitivity Troponin T (hsTNT), reflecting cardiac injury, however, were found in about a quarter of TCS in younger PWE undergoing VEM [62]. Intriguingly, none of the patients in this prospective clinical study had apparent cardiac complaints and no cardiac diseases were known or found in those with elevated hsTNT. Instead, the hsTNT levels were associated with high dopamine levels, indicating that the release of catecholamines and strong sympathetic activation were involved. In contrast, clinical TCS features were not linked to signs of cardiac injury [62].
These observations suggest that asymptomatic increases of cardiac injury markers in association with epileptic seizures can be interpreted as a mild form of neurogenic stress (Takotsubo) cardiomyopathy. Takotsubo cardiomyopathy is linked to very high catecholamine levels that induce cardiac contraction band necrosis [67]. The typical features of Takotsubo cardiomyopathy include chest pain, elevated troponin levels, transient ventricular motion abnormalities, and ECG alterations, similar to those of acute myocardial infarction in the absence of CAD [68]. The symptoms are commonly reversible, and the prognosis is mostly favourable. In the setting of emergency departments, elevations of troponin levels (cardiac troponin I or hsTNT) were found in 6-12% of TCS, especially in patients of older age (70 years or older), with cardiovascular risk factors or CAD [69-72]. Acute myocardial infarctions were only diagnosed in 2.2% of the patients referred to emergency departments because of TCS [69, 72]. Takotsubo cardiomyopathy can occur in the context of all forms of cerebral injuries, but in PWE, it usually occurs in association with TCS or status epilepticus [73].
Altogether, these findings underscore that troponin elevations linked to TCS need to be considered in greater detail. In the absence of CAD and in younger patients without cardiovascular risk factors, the TCS-related catecholamine release may lead to neurogenic stress without cardiac complaints or clinically manifest as Takotsubo cardiomyopathy, whereas in the elderly or patients with CAD, TCS may cause myocardial infarction.
How to manage seizure-related cardiac complications
Seizure-related arrhythmias may be recorded incidentally during VEM or shortly following a TCS after admission. Clinical clues that should prompt further investigations include onset of new seizure-related symptoms such as LOC that differ from the usual loss of awareness and unusual (atonic) falls. Seizure freedom will help to prevent seizure-related arrhythmias but cannot always be achieved. If refractoriness is already proven and epilepsy surgery not feasible, implantation of a cardiac pacemaker in patients with ictal bradyarrhythmia and a defibrillator in those with ventricular tachyarrhythmia is advisable. In PWE of all ages with new chest pain, with known CAD or in patients older than 70 years, it is advisable to determine troponin levels and perform a 12-lead ECG after TCS to exclude myocardial infarction and Takotsubo cardiomyopathy (table 4).
Epilepsy-related cardiac complications
Epilepsy is associated with a three-fold increased risk of sudden cardiac arrest, compared to the general population [5]. Importantly, about two thirds of the witnessed cases of sudden cardiac arrest in PWE were reported without evidence of a habitual seizure prior to the cardiac event, indicating that potentially lethal cardiac arrhythmias do not necessarily require epileptic seizures as a proximate trigger [74]. Hence, it is important to identify interictal cardiac findings in PWE that may predispose to SCD. These interictal epilepsy-related alterations, in turn, may be related to the functional or structural origin in the brain (central autonomic nervous system) or heart, and may be due to cerebral damage or specific localisation of the seizure-generating networks, intrinsic or genetic factors (e.g. inherited channelopathies), effects of antiseizure medication (ASM; see section Treatment-related cardiac complications) or remote effects of recurrent seizures.
Cardiac autonomic dysfunction
There is unequivocal evidence that interictal cardiac autonomic function is altered in PWE. For instance, interictal sympathetic tone tends to be enhanced whereas parasympathetic tone appears impaired, resulting in decreased heart rate variability (HRV) [75-77]. In non-epilepsy populations, decreased HRV is associated with elevated cardiovascular mortality [78]. Decreased HRV is found to be most severe in those with temporal lobe epilepsy and people with difficult-to-treat epilepsies [76]. The origins of autonomic dysregulation in epilepsy are unclear, but it is likely that brain areas controlling cardiac autonomic function are functionally or structurally affected by the seizure-generating networks, while peripheral components of the autonomic nervous system might also be affected during the course of the disease.
The ‘epileptic heart’
The duration of epilepsy appears to be linked to the development of cardiac diseases, as the prevalence of heart diseases is higher in PWE compared to people without epilepsy at all ages, and the age-related increase tends to be greater in PWE [79]. This observation supports the hypothesis that cardiac damage is incrementally acquired due to epilepsy-related autonomic dysfunction and to effects of recurrent seizures with myocardial catecholaminergic toxicity, as well as subtle ischaemic injury of cardiomyocytes and small cardiac nerves fibres [80]. Epileptic seizures, especially TCS, lead to sustained hypoxemia in the presence of strong and non-physiological generalized muscle activity, accompanied with a massive release of catecholamines, altogether leading to release of inflammatory factors and metabolic alterations [62, 81, 82]. This, in turn, causes cardiac stress even in the absence of acute cardiac complaints, as documented by asymptomatic release of hsTNT [62]. The structural correlate of such repetitive cardiac injuries may be cardiac fibrosis, which is found in about one third of PWE in post mortem analysis [13, 83].
In line with these observations, Verrier and colleagues have recently proposed the concept of the ‘epileptic heart’, defined as “a heart and coronary vasculature damaged by chronic epilepsy as a result of repeated surges in catecholamines and hypoxemia leading to electrical and mechanical dysfunction” [13]. Structural and functional heart properties can also be assessed at a more macroscopic level by echocardiography, and specific findings of systolic and diastolic dysfunction or hypertrophy are linked to an increased SCD risk [84, 85]. In adults with temporal lobe epilepsy, left ventricle stiffness was increased with higher pressure compared to healthy controls, which was probably related to altered cardiac autonomic activity and possibly cardiac fibrosis, promoting systolic and diastolic dysfunction and arrhythmogenesis [86]. Echocardiographic studies in children with epilepsy have also shown greater thickness of carotid intima-media and epicardial adipose tissue in epilepsy versus non-epilepsy (particularly in those epilepsy patients on polytherapy vs monotherapy), left ventricle myocardial deformation and impaired systolic and diastolic functions in children with ASMs and without seizures in the last six months versus healthy controls, and left ventricle function deterioration (contraction time, relaxation time, performance index and ejection time) in untreated ‘idiopathic epilepsy’ versus healthy controls [87, 88].
What are the potential consequences of this incremental damage? Subtle perivascular and interstitial fibrotic microlesions in the heart could contribute to or cause disturbed conduction of cardiac excitation, heterogeneous cardiac repolarisation and heterotopic excitation, thereby serving as potential sites for the generation and maintenance of fatal arrhythmias such as VT/VF. Apart from (micro-)structural damage of the heart, emerging data from animal studies of acquired epilepsies also demonstrate changes of cardiac ion channel expression levels (e.g. SCN1A, SCN5A, HCN2) that translate into altered electrical properties of the heart, strengthening the notion of the ‘epileptic heart’ [89].
Altered cardiac repolarisation indices
Taken together, cardiac microlesions, altered expression of cardiac ion channels and enhanced sympathetic tone may also favour abnormally altered cardiac repolarisation features (table 1) and onset of potentially lethal VT/VF [90]. Indeed, interictal prolongation of QT intervals (corrected for heart rate [QTc]) is more common in PWE compared to people without epilepsy [6, 91-94]. Some studies reported similar QTc durations in both people with and without epilepsy [95, 96], while others found QTc shortening [97, 98]. Older PWE and those with difficult-to-treat epilepsies, in particular, are at higher risk of development of QTc prolongation compared to people without epilepsy [6, 94]. Furthermore, ventricular late potentials were found in 22 of 45 PWE compared with one of 19 controls [99], and QT dispersion, as a measure of regional heterogeneity of cardiac repolarization, was reported in up to one third of PWE [18]. The latter may be of particular interest, because abnormal QT dispersion is thought to facilitate onset of VT/VF when abnormal QT shortening occurs, which could be a possible explanation for onset of VT/VF in association with TCS [90].
T wave alternans (TWA) is another marker of cardiac repolarisation which has recently gained attention in the field of epileptology [100]. TWA is the beat-to-beat fluctuation of morphology and amplitude of the ST segment or T wave, reflects temporal-spatial heterogeneity of cardiac repolarization and predicts cardiovascular mortality and SCD in people with cardiovascular disease [101]. The higher the level of TWA, the more likely is the onset of ventricular tachyarrhythmia [101]. TWA values in PWE were found to be elevated above the 47-μV abnormality cut-off point in most patients (82-100%) [102, 103]. Furthermore, TWA levels appear to depend on the duration of epilepsy, again supporting the notion of acquired cardiac damage. People with chronic epilepsy displayed higher TWA levels than those with newly diagnosed epilepsy, and TWA levels of adults with newly diagnosed epilepsy did not differ from healthy controls [104]. Importantly, PWE had reduced TWA levels and increased HRV upon vagus nerve stimulation (VNS), suggesting that VNS can considerably reverse or compensate epilepsy-related cardiac dysfunction [102, 103]. A prospective study in subjects with chronic heart failure supports the notion that VNS has cardioprotective effects, as those treated with VNS had reduced TWA levels, improved baroreflex sensitivity and fewer episodes with VT [105]. Whether these effects (partially) explain why long-term VNS treatment reduces SUDEP risk, however, remains to be elucidated [106].
Inherited cardiac dysfunction
In addition to acquired cardiac diseases, PWE with sudden cardiac arrest due to VT/VF were also shown more often to have pre-existing congenital or inherited heart diseases than controls [64]. Genetic defects may cause a multiple malformation syndrome affecting the development of the heart and brain or congenital heart disease, or abnormal cardiovascular function may lead to poor (intrauterine) brain growth. Alternatively, ion channel mutations may induce both cardiac arrhythmias and epilepsy. For instance, severe QTc prolongation and certain epilepsy syndromes are associated with sodium and potassium channel mutations. An ion channel mutation expressed in brain and heart, a cardiocerebral channelopathy, might lead to a propensity for seizures and cardiac arrhythmias. Numerous cardiocerebral channelopathies are known including pathogenic variants in the long QT gene family (i.e. SCN5A, KCNQ1 and KCNH2) that are associated with epilepsy (i.e. self-reported diagnosis and ASM use) [107-112]. Cardiac channelopathy genes not associated with long QT, such as RYR2 and HCN1-4, may predispose to epilepsy, as indicated by mouse model studies [113-116]. Likewise, gene mutations typically associated with epilepsy such as SCN8A, CDKL5, CNTNAP2 and GRIN2A may also cause cardiac arrhythmias, further underscoring the concept of genetic pleiotropy and cardio-cerebral channelopathies [117, 118]. Mutations in some of these cardio-cerebral genes were also found in people who died suddenly, stressing the potential contribution of cardio-cerebral phenotypes to SUDEP [117, 119].
How to manage epilepsy-related cardiac complications
A 12-lead ECG is advisable in all patients at the time of first epilepsy diagnosis to identify constellations suggestive of inherited cardiac arrhythmia (e.g. long / short QT syndromes) and to mitigate the risk of false diagnosis of epilepsy. Given the possible presence of both entities, i.e. acquired or genetically caused epilepsy and cardiac arrhythmias, one should be aware that the presence of abnormal QT intervals on 12-lead ECG does not exclude epilepsy. Further examinations, e.g. prolonged EEG recordings, may be required if (additional) epilepsy is suspected. In PWE who report new onset of palpitations, episodes with LOC in the absence of typical seizure symptoms or other cardiac complaints, a 12-lead ECG should be performed. If treatment-related causes of cardiac complications are excluded (see next section), referral to a cardiologist for further examinations (Holter recordings, echocardiography, implantation of a loop recorder) is recommendable (table 4).
Treatment-related cardiac complications
PWE chronically receive ASM or neuromodulatory devices including VNS. Given the broad spectrum of direct seizure- and epilepsy-related cardiac effects in the continuous presence of antiseizure therapies, it can be challenging to identify treatment-related cardiac complications.
Cardiac complications of antiseizure medication
ASM seem to be an independent risk factor for SCD, especially in patients taking sodium channel blocking agents, even in the absence of epilepsy [16, 120]. Many ASM predominantly act on excitatory voltage-gated sodium channels, but may also bind to various other targets. For instance, carbamazepine, lamotrigine and phenytoin which are considered as classic sodium channel blockers also inhibit calcium channels, whereas levetiracetam appears to mediate its antiseizure effects via binding to the presynaptic synaptic vesicle protein 2A, but also acts on calcium channels [121]. Importantly, voltage-gated sodium and calcium channels are expressed both in the brain and heart with distinct preferential expression patterns according to the channel type. Since binding affinities are not absolute for “brain channel subtypes”, ASM might lead to cardiac dysfunction and cardiac arrhythmias indirectly via modulating the central autonomic nervous system and via direct binding to cardiac tissue. Preclinical data have also shown that some ASM (lamotrigine, phenobarbital, phenytoin) inhibit the cardiac rapid-delayed rectifier potassium ion current [122, 123], which could cause prolongation of QT intervals and possibly facilitate VF/VT [124]. In this context, one may also discuss safety issues of ASM in epilepsy populations with inherited and acquired cardiac disease, as data are lacking on binding affinities and effects of ASM on mutant cardiac channels associated with arrhythmias. Indeed, in a recent cohort study on people with genetic long QT syndromes, the use of ASM was associated with an increased risk of cardiac events, especially in people with long QT syndrome type 2 [125]. Cardiac events were more frequent in female patients and upon sodium channel blocking agents, whilst co-administration of beta-adrenergic blocker medication attenuated the risk of cardiac events [125].
Given these considerations, one might expect frequently occurring new-onset cardiac arrhythmias in PWE after starting e.g. “sodium channel blockers”. In the randomized controlled trials for drug approval and additional safety studies of the different sodium channel blocking agents, however, most ASMs were shown to be safe (and those drugs with strong effects on cardiac conduction and repolarisation were not approved). In line with this, ASMs usually do not exert clinically relevant effects on cardiac repolarisation properties and QT intervals [90]. In the case of pre-existing conduction abnormalities (e.g. higher-grade AV-block), however, sodium channel inhibitors may be contraindicated, as the risk of deteriorating the conduction deficits to a full block is elevated. Importantly, at very high doses, after ingestion (“overdose”) or in combination with other sodium channel blocking agents or ASMs at higher daily doses, sodium channel inhibitors were reported to cause cardiac arrhythmias, independently of pre-existing cardiac diseases. The use of carbamazepine, lacosamide, lamotrigine and phenytoin was mostly associated with bradyarrhythmias (such as sinus node dysfunction and AV block), but also with ventricular tachyarrhythmia and atrial fibrillation [126-131].
The reports on ASM-induced alterations of cardiac autonomic control and HRV are controversial, but according to a meta-analysis including six original studies, there is no apparent effect of ASM on HRV [132]. In contrast, ASM intake and poor circulating cardiovascular risk factors were shown to be unequivocally linked, especially with enzyme-inducing ASMs (i.e. carbamazepine, phenytoin, phenobarbital, primidone) which worsen the lipid profile and other serological markers associated with vascular risk via induction of the hepatic cytochrome P450 system [133, 134]. These alterations, in turn, favour accelerated atherosclerosis, reflected, for instance, by the finding that duration of intake of enzyme-inducing ASMs was associated with the thickness of the intima media of the common carotid artery, an established vascular risk marker [135]. Some ASMs (e.g. gabapentin, pregabalin and valproate) are also associated with weight gain, which increases the risk of developing non-alcoholic fatty liver disease and metabolic syndrome, thereby further worsening cardiovascular risks [15].
Cardiac complications of vagus nerve stimulation
VNS involves a device that is implanted under the skin of the chest which is connected to the vagus nerve in the mid-cervical region. The device delivers periodically (e.g. every five minutes) an electrical current lasting for 30 seconds to the vagus nerve. In PWE, the left vagus nerve is usually chosen because it predominantly contains afferent fibres projecting to the brain and less efferent fibres projecting to the heart, thereby mitigating cardiac complications and enhancing potential CNS effects. Little is known about the exact mechanisms leading to improved seizure control upon VNS, but these are likely to involve significant changes of neurotransmitter levels in the brain [136], leading to a reduction in seizure frequency of at least 50% in 30 up to 50% of PWE [137]. Newer VNS devices can also apply additional pulses upon a seizure-related increase in heart rate, as detected by a closed loop system, which may interrupt seizure activity and shorten seizure duration [138].
The reports on cardiac autonomic control and HRV upon VNS are mixed, and a meta-analysis including five studies has not revealed a clear effect of VNS on HRV [132]. One might expect an elevated risk of bradycardia and asystole upon VNS, but because of the implantation of the left vagus nerve (with few fibres projecting to the AV node only) and the systematic testing of cardiac effects during the implantation procedure, VNS-induced cardiac complications are very rare. Indeed, only a few cases with delayed onset (several years after VNS implantation) of episodic bradycardia and syncope during the on-phase of the VNS were reported [139]. It is important to note that cholinergic innervation (by vagus nerve fibres) is not only present in the sinoatrial and AV node, but also in the human ventricular conducting system, allowing modulation of electrical properties of the cardiac ventricles [140]. Indeed, TWA levels were significantly reduced upon VNS (see also the section Epilepsy-related cardiac complications), suggesting that VNS stabilizes electrical properties of the heart and possibly mitigates the risk of VT/VF and SCD [102, 103]. This, however, needs to be confirmed in larger populations.
How to manage treatment-related cardiac complications
ASMs are required for antiseizure treatment. If successful, the use of ASMs prevents seizure-related cardiac arrhythmias and remote cardiac complications (‘epileptic heart’). However, enzyme-inducing ASMs should be avoided at least in those with cardiovascular risk profiles, as these drugs may aggravate cardiovascular risk factors including weight gain linked to specific ASMs. In patients with new onset of syncope or episodes of TLOC in the absence of habitual seizure-related symptoms, a 12-lead ECG (or Holter recordings) is advisable to search for ASM-induced arrhythmias. If a modification of antiseizure drug treatment involving sodium channel blockers is intended, a 12-lead ECG (before and e.g. two weeks after the modification), to exclude AV block or sinus node dysfunction, is advisable for the following constellations: patients with other cardioactive substances (e.g. antihypertensive or antiarrhythmic drugs), or in those with high doses or a combination of sodium channel blockers. Rare episodic bradycardia or asystole should be considered in patients with VNS if they report onset of new symptoms or LOC in the absence of typical seizures (table 4).
Supplementary data
Supplementary didactic slides are available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
PR has received speaker or consultant fees from UCB pharma, Eisai pharmaceutical, LivaNova and GW pharmaceuticals. RS has received speaker fees or honorary for serving on the advisory board for Bial, Desitin, Eisai, LivaNova, Novartis and UCB Pharma, and research support from the Federal Ministry of Health, Federal Ministry of Education and Research, and the Boll foundation (Kerpen, Germany). RDT receives research support from Medtronic, Dutch National Epilepsy Fund, The Netherlands Organization for Health Research and Development (ZonMW), Christelijke Vereniging voor de Verpleging van Lijders aan Epilepsie NUTS Ohra Fund, and the AC Thomson Foundation, and reports consultancy and lecturing fees from Theravance Biopharma, Medtronic, UCB and Novartis and research support from Medtronic.CD and SS report no disclosures.
References for further reading
van Dijk JG, Thijs RD, van Zwet E, Tannemaat MR, van Niekerk J, Benditt DG, et al. The semiology of tilt-induced reflex syncope in relation to electroencephalographic changes. Brain 2014; 137: 576-85.
Palma JA, Benarroch EE. Neural control of the heart: recent concepts and clinical correlations. Neurology 2014; 83: 261-71.
Van der Lende M, Surges R, Sander JW, Thijs RD. Cardiac arrhythmias during or after epileptic seizures. J Neurol Neurosurg Psychiatry 2016; 87: 69-74.
Brigo F, Lochner P, Nardone R, Manganotti P, Lattanzi S. Increased risk of stroke and myocardial infarction in patients with epilepsy: a systematic review of population-based cohort studies. Epilepsy Behav 2020; 104: 106307.
Mintzer S, Skidmore CT, Abidin CJ, Morales MC, Chervoneva I, Capuzzi DM, et al. Effects of antiepileptic drugs on lipids, homocysteine, and C-reactive protein. Ann Neurol 2009; 65: 448-56.
Key points
- •People with epilepsy have a 2-3-fold increased risk of dying prematurely, which is due to sudden cardiac death or acute myocardial infarctions in a considerable proportion of patients.
- •Cardiac complications in people with epilepsy can be caused by epileptic seizures, treatment, or epilepsy itself.
- •Syncope can be mistaken for epileptic seizures, which hampers the timely diagnosis of cardiac complications in people with epilepsy or leads to false diagnosis in people without epilepsy.
- •Ictal asystole is the most frequent clinically relevant arrhythmia, occurs predominantly with focal seizures in temporal lobe epilepsy, has a high recurrence risk, and may require cardiac pacemaker implantation if seizure freedom cannot be achieved.
- •Seizure-related ventricular tachycardia is rarely reported and only in the context with tonic-clonic seizures.
- •Seizure-related myocardial infarction is rare and predominantly occurs with tonic-clonic seizures in elderly patients or in those with known coronary artery disease.
- •Two thirds of sudden cardiac arrests in people with epilepsy are reported in the absence of seizures, indicating that potentially lethal cardiac arrhythmias do not require epileptic seizures as a proximate trigger.
- •Epilepsy-related interictal cardiac complications include altered cardiac autonomic function with decreased heart rate variability and abnormalities of cardiac repolarisation features, possibly facilitating onset of ventricular tachyarrhythmias.
- •Antiseizure medication may prevent seizure-related cardiac arrhythmias and remote cardiac complications (‘epileptic heart’).
- •Enzyme-inducing antiseizure medication should be avoided at least in those with cardiovascular risk profiles, as these drugs may aggravate cardiovascular risk factors including weight gain linked to specific ASMs; sodium channel blockers at very high doses or in combination can lead to potentially life-threatening cardiac arrhythmia, mostly bradyarrhythmias, but also atrial and ventricular tachyarrhythmias.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License