Epileptic Disorders
MENUHow to distinguish seizures from non-epileptic manifestations Volume 22, issue 6, December 2020




Learning objectives and competencies:
1.3 Identify and describe seizure semiology using standardized ILAE terminology and classification systems
1.3.1 Learn and recognize the seizure semiology and distinguish it from other non-epileptic manifestations:
- •Recognize characteristics typical of epileptic seizures (L1)
- •Recognize features typical of psychogenic non-epileptic seizures (PNES) (L1)
- •Recognize characteristics of other paroxysmal events in adults (L1)
- •Recognize characteristics of other paroxysmal events in newborns, infants, children and adolescents (L1)
According to The International League against Epilepsy (ILAE), an epileptic seizure (ES) is defined as “transient occurrence of signs/symptoms because of abnormal excessive or synchronous neuronal activity in the brain” (Fisher et al., 2005). The clinical presentation of an ES is diverse and dependent upon the involved regions of the cerebral cortex. Semiological features comprise motor and non-motor phenomena (sensory, autonomic, behavioural change, cognitive, emotional) with or without impairment of awareness (Fisher et al., 2017). These symptoms also occur in several other conditions which may mimic seizures. In adults, the most common imitators of epilepsy are syncope and psychogenic non-epileptic seizures (PNES), followed by migraine, parasomnias, cerebrovascular disease including transitory ischaemic attack (TIA), and movement disorders such as paroxysmal dystonia and non-epileptic myoclonus (Scheepers et al., 1998; Benbadis, 2009; Xu et al., 2016). In the paediatric population, differential diagnoses of ES includes numerous conditions and behavioural states. For the clinical diagnosis of epilepsy, identification of epileptic seizures is crucial. Hence, this review focusses on the core competencies of Chapter 1.3.1 of the ILAE Epileptology Curriculum (Blümcke et al., 2019): Learn and recognize the seizure semiology and distinguish it from other non-epileptic manifestations. These core competencies are relevant to all clinicians diagnosing and managing people with epilepsy.
Diagnosis of epilepsy starts with a comprehensive history, identifying characteristics to aid in distinguishing seizures from other paroxysmal disorders. The inter-ictal neurological examination may be normal or may show focal abnormalities. The ictal semiology is one of the key components to differentiate epileptic from non-epileptic manifestations. Diagnostic tests, including EEG, MRI, blood test or neuropsychological assessment, will support the diagnosis of seizures but may be normal in patients with epilepsy. However, tests may reveal non-specific findings, which can be erroneously interpreted as supporting the diagnosis of epilepsy. This underlines the importance of clinical factors in the diagnostic process (Angus-Leppan, 2008). Aspects regarding diagnostic tests are covered in other papers of this series.
Accurate diagnosis is paramount in order to ensure the most appropriate management at an early stage of the disease. In 20-30% of adults who are diagnosed with epilepsy, the diagnosis of epilepsy is incorrect. Misdiagnosis rates are even higher in patients with epilepsy refractory to therapy (Xu et al., 2016). However, consultation with specialists increases diagnostic accuracy (Scheepers et al., 1998; Leach et al., 2005; Angus-Leppan, 2008).
Diagnostic uncertainty is due to several reasons. Firstly, as mentioned above, there is a significant overlap of clinical features between other conditions. Secondly, the level of physicians’ clinical expertise may be a contributory factor. And finally, a lack of accurate history of patients and descriptions based on witnesses may pose a diagnostic challenge (Smith, 1999; Chowdhury et al., 2008; Syed et al., 2011).
Studies examining the description of seizures based on reports from caregivers reveal that what is reported by family members may not reflect the actual ictal semiology. In a study of video-EEG-documented ES and PNES, 120 seizures (84 ES, 36 PNES) were analysed from 35 consecutive patients (Syed et al., 2011). Only three ES signs (“abrupt onset”, “eye-opening/widening”, and postictal “confusion/sleep”) and three PNES signs (“preserved awareness”, “eye flutter”, and “bystanders can intensify or alleviate”) were significant and reliable indicators of seizure type from among 45 video-documented signs. Of note, eyewitness reports of these six signs were inaccurate and not statistically different from guessing. The authors found that eyewitness reports of signs did not predict video-EEG-ascertained diagnosis. Studies indicate an average delay in making a definite diagnosis of psychogenic non-epileptic seizures (PNES) of over seven years.
However, regardless of the underlying correct diagnosis, misdiagnosis of epilepsy leads to a significant lag of sufficient treatment and conversely to inadequate therapy, such as prescription of antiepileptic drugs (AEDs) (Kerr et al., 2016; Bahrami et al., 2019). Side effects and potential teratogenic effects of AEDs must not be disregarded (Chadwick and Smith, 2002; Chowdhury et al., 2008; Xu et al., 2016). Further consequences and risks including social implications on patients’ lifestyle, employment and driving, associated with a false positive diagnosis of epilepsy, have to be considered (Oto, 2017). Table 1 outlines the most common semiological features that occur in epilepsy and the most frequent differential diagnoses in adults.
Paroxysmal non-epileptic manifestations in the paediatric age group constitute a complex group of conditions, including recurrent intermittent motor movements, behavioural changes and somatic symptoms. Table 2 shows a list ofparoxysmal non-epileptic events/disorders occurring under 18 years, sorted by age.These may be associated with different signs and symptoms including fainting, loss of consciousness, headache, vomiting, dizziness, abdominal pain, irregular breathing, sleep disorders, and emotional and psychiatric problems. These manifestations typically present repetitively with a sudden onset and end. They may last for seconds or minutes and generally occur at specific ages, and most importantly, similarly in adults, may be misdiagnosed as epilepsy and treated unnecessarily.
Consequently, the first step in the diagnostic process of a patient in any age group presenting with a paroxysmal neurological event is an attempt to answer the question: “is it a seizure or not?”. In order to distinguish ES from other entities, characteristics of semiology, including signs preceding the event, onset/offset, duration, frequency, type, sequence and continuity of ictal behaviour, are considered (Vogrig et al., 2019). Attention should also be paid to precipitating factors, e.g. emotion, anger, tiredness, happiness, auditive/sensory stimulation, the circumstances of the attacks, e.g. in sleep or in the awake state or involving a specific posture (sitting, walking, in a high chair or sitting in a car, or in a vertical position), and on any triggers which cause the attacks to terminate. Further, the presence or absence of prodromes and the postictal period may be helpful to differentiate between conditions (Izadyar et al., 2018). The postictal period is defined as an abnormal state that may be characterized by impaired awareness, motor or neuropsychological functions that last from the end of the ictal phase until the return to the presumed baseline (Rémi and Noachtar, 2010).
Given the diverse specificity and sensitivity of semiological signs and the variability in accuracy among different observers, diagnosis should not solely rest on one single feature (Avbersek and Sisodiya, 2010; Seneviratne et al., 2012). Home videos have facilitated the diagnosis significantly (Tatum et al., 2020). However, in some cases, recording of video-EEG may be necessary. Correct diagnosis can prevent unnecessary investigations and medical treatment.
In the following sections, the main features of epileptic seizures and non-epileptic episodes for both, adults and children, are discussed.
Adults and adolescents
Features of epileptic seizures (ES)
Typical of ES (but not limited) is an abrupt onset, a duration of less than two minutes in accordance with a self-limiting process except for status epilepticus, and a prolonged postictal state, which depends on seizure type and origin.
Precipitating features
Triggers for ES among patients with epilepsy include missing AED medication, emotional stress, sleep deprivation, fatigue, alcohol intake and fever (Balamurugan et al., 2013; Ferlisi and Shorvon, 2014). ES may be further provoked through metabolic disturbances (i.e. hyponatraemia, hypoglycaemia), drug or alcohol withdrawal, or toxic or medication side effects (Beghi et al., 2010). Moreover, it is important to keep in mind that recent events in the CNS such as stroke, infection or traumatic brain injury may cause ES (Bergey, 2016).
Features of focal seizures
A possible clinical key feature of ES is the presence of a typical aura. According to the latest classification of seizures by the ILAE the term “aura” was replaced by “focal aware seizure with sensory or emotional (non-motor) onset” (Fisher et al., 2017). Given the different clinical features of an aura, which may mimic other conditions such as migraine, the time course and associated symptoms may help to differentiate between ES and other entities. Typically, aura symptoms tend to appear in seconds and might include propagation, adding additional symptoms. The development of more obvious seizure symptoms or symptoms typically associated with other diseases (i.e. headaches, migraine) throughout the event may facilitate diagnostic identification (Benbadis, 2009).
The occurrence of déjà vu experiences combined with other features, such as phenomenon of derealisation or an epigastric aura, is highly specific to ES (Adachi et al., 2006, 2010). By contrast, the isolated appearance of déjà vu is considered as a non-epileptic event, as it occurs in patients with neuropsychiatric disorders or physiologically normal individuals (Adachi et al., 2006, 2010).
Focal seizures are otherwise often characterized by an abrupt onset of neurologically, mostly positive symptoms, such as tonic or clonic movements, automatisms, abnormal sensations or hallucinations. Later, during the course of the seizure, symptoms disappear when the seizure ends or spread with potential change in symptoms according to the propagation of abnormal electrical activity in the brain (Berg et al., 2010). A typical example of such propagation is the march of clonic movements through different parts of the affected side of the body in focal motor seizures or progression of a focal aware seizure with epigastric sensation into a focal unaware seizure with automatisms. Patients with ES may also experience negative ictal symptoms. These include blindness in occipital epilepsy, negative myoclonus, aphasia, and amnesia (Manford, 2001). The duration of the ictal phase of focal seizures is usually significantly shorter compared to bilateral tonic-clonic seizures (BTCS), varying from seconds to two minutes (Dobesberger et al., 2015). The exact description of different types of focal semiology is beyond the scope of this paper and covered elsewhere.
Convulsions
In bilateral tonic-clonic seizures (BTCS; previously “generalised tonic-clonic seizures”), a certain sequence of symptoms typically occurs which is composed of a tonic posturing phase followed by a clonic phase with rhythmic body movements (Berg et al., 2010). Typically, fast jerks with low amplitude evolve to slower, higher-amplitude jerks. Focal ictal symptoms may precede the generalised phase of the seizures in focal to bilateral tonic-clonic seizures (Fisher et al., 2017). The classic guttural epileptic “cry” in epilepsies of generalised origin is a unique sound that is essentially pathognomonic for BTCS (Elzawahry et al., 2010).
Duration
The ictal period usually lasts 1-3 minutes. Focal seizures are usually of shorter duration, with a minimum duration of a few seconds in some frontal lobe seizures. BTCS tend to last on average less than 2 minutes. The character of self-limitation is lacking if BTCS or focal seizures last longer than 5 or 10 minutes, respectively, or if BTCS recur without regaining consciousness within 30 minutes, fulfilling the criteria for status epilepticus (Trinka et al., 2015).
Eye opening
Eyes are usually open in ES with gaze straightforward or deviated (Chung et al., 2006; Syed et al., 2008; LaFrance Jr. et al., 2013). Since eye-opening behaviour can also be clearly observed by witnesses or recorded on video documentation, it is suggested as a good clinical sign in order to distinguish ES from PNES in particular. Eye opening or widening at onset, abrupt onset and postictal-confusion or sleep are reported as the best semiological predictors of ES (Syed et al., 2011).
Stereotypic features
Regarding stereotypic features, the formerly common belief was that these predominantly occur in ES. However, stereotypic behaviour is also observed in PNES, and is therefore no longer considered a reliable factor to distinguish between ES and non-epileptic seizures (Seneviratne et al., 2010; Herskovitz, 2017).
Incontinence, injury and tongue biting
Contrary to the notion that certain common signs are specific to epilepsy, incontinence, significant injury and tongue lacerations occur in both ES and PNES (Desai et al., 1982; Guberman, 1982; Hoefnagels et al., 1991; Meierkord et al., 1991; Benbadis et al., 1995). At least one of the typical signs associated with BTCS (e.g., tongue biting, falling or urinary incontinence) was reported by 66% of patients with PNES (de Timary et al., 2002), therefore their presence does not necessarily indicate that the event is epileptic. Obviously, documented incontinence or tongue biting are much more specific than reported incontinence or tongue biting. Lateral tongue biting is highly specific to BTCS, and thus is a very helpful sign for ES when present (Benbadis et al., 1995; Dufresne et al., 2019). In a systematic review based on pooled analyses of data from the literature, urinary incontinence was reported to be of no value as either a diagnostic sign itself or a discriminating feature between ES and PNES/syncope (Brigo et al., 2013a).
Postictal recovery
The postictal period in focal and generalized seizures usually lasts less than 24 hours, and its duration may vary from seconds to days, and rarely months. The median duration of the postictal state after a bilateral tonic-clonic seizure is 45 minutes (Ohira et al., 2019). Patients with older age, longer seizure duration and impaired brain function at baseline experience an even longer postictal state (Kellinghaus et al., 2004; Ohira et al., 2019), which may lead to misdiagnosis of dementia (DeToledo, 1999; Theodore, 2010; Subota et al., 2019). The longest postictal period was reported in patients with cognitive and behavioural symptoms, lasting close to two months (Subota et al., 2019). A very short duration or complete lack of postictal phase is atypical of BTCS, but well known in focal frontal lobe seizures with preserved awareness or generalized non-motor seizures with impaired awareness (previously “absence seizures”).
The postictal period may be associated with various physiological and behavioural symptoms, including postictal headaches, Todd's paresis, aphasia, coughing, nose rubbing, psychosis and affective symptoms (Gallmetzer et al., 2004; Hoffmann, Elger and Kleefuss-Lie, 2009; Rémi and Noachtar, 2010; Wang et al., 2014; Gavvala et al., 2015). Postictal paresis is reported to occur in 10% patients with focal epilepsy, lasting for more than 2 minutes to 36 hours. Postictal aphasia is experienced in 34% (Subota et al., 2019). Post-ictal nose rubbing and coughing was observed after focal epileptic seizures, but not after BTCS or PNES or other entities (Geyer et al., 1999). The mechanisms involved are barely understood, but may result from lower neuron excitability in brain regions involved in the seizure activity (Fisher and Schachter, 2000). Further, it was stated, that postictal automatisms, such as kissing, biting and nose wiping, may represent a release phenomenon (Tassinari et al., 2005; Rashid et al., 2010; Rémi and Noachtar, 2010).
A postictal breathing pattern after BTCS has been described as stertorous, i.e. deep, loud and regular (similar to gurgling and snoring in deep sleep) (Sen et al., 2007; Azar et al., 2008; Rosemergy et al., 2013). This results from ictal hypersalivation leading to saliva being whisked in the throat by the clonic breathing pattern (foaming at the mouth) (Azar et al., 2008).
Psychogenic non-epileptic seizures (PNES)
Psychogenic non-epileptic seizures (PNES) are characterized by paroxysmal movements, sensations, or experiences that may resemble epileptic manifestations, are not associated with epileptic discharges on the EEG (Metrick et al., 1991), and are generally associated with underlying psychological stressors. PNES are among the most frequent non-epileptic paroxysmal manifestations in adolescents, but may occur at any age including children younger than 10 years (Kutluay et al., 2010) and in the elderly. When observing ictal signs, it is essential to understand that no single characteristic is pathognomonic for PNES. Certain characteristics of the motor phenomena, however, are strongly associated with PNES and are rare in ES, and are therefore relatively specific. When the typical PNES includes more than one of these signs, which is common, the diagnosis of PNES is more likely. Notably, differential diagnosis can be challenging due to the 10% comorbidity of epilepsy and PNES (Benbadis et al., 2001). Hence, careful evaluation of any new seizure type in a patient is required, and video-EEG monitoring might be necessary for correct diagnosis.
Precipitating factors
While epileptic and nonepileptic seizures can be “triggered” by an environmental stimulus, certain events may precipitate PNES, such as discussing a trauma history.
Motor and non-motor signs
“Pseudosleep” and discontinuous (stop-and-go), irregular or asynchronous (out-of-phase) activity, including side-to-side head movement, pelvic thrusting, opisthotonic posturing, stuttering, and weeping are behaviours or signs strongly suggestive of PNES (Desai et al., 1982; Guberman, 1982; Gulick et al., 1982; Gates et al., 1985; Meierkord et al., 1991; Bergen and Ristanovic, 1993; Vossler et al., 2004; O’Sullivan et al., 2007). The atypical motor features of PNES can be objectively measured and quantified with techniques such as EMG (Beniczky et al., 2014) or time-frequency mapping (Bayly et al., 2013). Furthermore, ictal behaviours that are modified by an examiner, such as avoiding a noxious stimulus (“self-protective manoeuvre” after dropping the hands of an unresponsive patient over his/her face), and non-anatomical symptom progression (various limbs moving at various times) can also help with seizure type differentiation. Another useful sign to indicate PNES is preserved awareness and ability to interact with the examinerduring bilateral motor activity.
Duration
PNES demonstrate a prolonged ictal duration (≥5 minutes). Seizure duration of over 5 minutes indicates a 24-fold higher likelihood of PNES diagnosis. The optimal threshold value to differentiate between PNES and ES is reported to be 123.5 seconds with a sensitivity of 65% and specificity of 93% (Seneviratne et al., 2017).
Eye closure
Ictal eye closure at onset is associated with PNES (Chung et al., 2006), and although this sign has been questioned (Syed et al., 2008), eye closure, especially when prolonged and with complete unresponsiveness, is relatively specific to PNES. Closed eyelids resisting passive opening are further indicative of PNES (Chung et al., 2006; Avbersek and Sisodiya, 2010; Syed et al., 2011; LaFrance Jr. et al., 2013).
Tongue biting and incontinence
As mentioned above, incontinence and injury may occur in ES or PNES. Whereas lateral tongue biting is more likely in ES, injury located at the anterior site of the tongue supports a diagnosis of syncope or PNES (Benbadis et al., 1995; Brigo et al., 2013b).
Postictal recovery
Postictal responses, such as whispering or partial motor responses, have a strong association with PNES (Chabolla and Shih, 2006). PNES are typically accompanied by shallow, quiet and irregular breathing characteristics in the postictal phase in contrast to stertorous breathing in ES (Azar et al., 2008). Rapid postictal recovery is common in PNES, whereas postictal “confusion” is widely known to follow most ES, however, the latter is noted by many patients with PNES and its absence does not rule out ES, as seen in certain frontal lobe ES and absence epilepsy.
Teaching medical observers specific signs to look out for may help distinguishing ES from PNES (table 3). In one study, nurses, emergency physicians, medical students and neurology residents were quizzed before and after being primed on distinguishing signs of ES and PNES, and all cohorts were found to improve diagnostic skills after the instructional programme (De Paola et al., 2016). Education on signs of PNES and ES may be helpful not only for clinicians, which may help with appropriate triage and intervention for the different seizure types, but also for office and hospital staff when events occur in the medical setting (LaFrance Jr and Wincze, 2015). Finally, teaching patients to observe their signs may impact seizures, and studies have shown that behavioural interventions with aura identification (Reiter et al., 2015) leads to a reduction in ES (Michaelis et al., 2012) and PNES (LaFrance Jr. et al., 2014).
Paroxysmal events
Syncope
Syncope may have a cardiac or neurocardiogenic origin. Initially, patients may report fading vision into black, thought to be caused by retinal hypoxia, often accompanied by dizziness and altered hearing. With asystole, consciousness is lost after 6-7 seconds. Eyes are open or half open, showing initial downbeat nystagmus, followed by upward gaze deviation. Loss of tone is seen in all syncope, whereas it is rarely seen in ES (Shmuely et al., 2018). Up to 80% of volunteers suffering with syncope after Valsalva manoeuvre showed convulsions (Lempert, 1996). These are irregular bilateral myoclonic jerks that include flexion of the elbows with supination of the wrist (Shmuely et al., 2018), lasting usually less than 30 seconds (Lempert, 1996). Hence, convulsive syncope is often misdiagnosed as BTCS, even by medical professionals. A recent publication postulated the 10/20 rule to differentiate syncope from ES. If a patient has less than 10 myoclonic jerks, syncope is more likely, and if a patient has more than 20, ES is favoured (Shmuely et al., 2018). Postictal recovery is usually quick, with the patient being fully orientated immediately or after a few minutes. Longer postictal recovery is described and may indicate additional or pure pseudo-syncope (Blad et al., 2015). As mentioned above, incontinence and tongue biting may occur during syncope; the latter typically involving the tip of the tongue. A list of semiological features, compared between ES, PNES and syncope, is presented in table 4.
Migraine
Migraine is a common neurological disorder with paroxysmal occurrence.
Migraine auras may mimic epileptic manifestations of a different nature, including occipital (visual symptoms), parietal (localized paraesthesia) and temporal (psychic phenomena) seizures. The development of more obvious seizure symptoms or typically migraneous headache throughout the event may facilitate diagnosis (Benbadis, 2009).
However, the high comorbidity of epilepsy and migraine (Lipton et al., 1994), the potential occurrence of headache-free aura episodes or the possibility of headache as the only ictal manifestation of an epileptic seizure (Parisi et al., 2015) may pose a diagnostic challenge. Some sensory features (paraesthesia or visual), such as slow, gradual evolution, are more in favour of migraine manifestations. Also, psychic phenomena related to migraine auras do not generally exhibit the strong emotional components associated with temporal lobe epileptic seizures. Typically, migraine aura symptoms evolve within minutes, whereas ES aura symptoms tend to appear in seconds. However, migraine with aura may also trigger an ES (Gowers, 1906). The third edition of the International Classification of Headache Disorders (Headache Classification Committee of the International Headache Society, 2018) lists migraine aura-triggered seizures as ES that occur during or within one hour of migraine with aura. The previously used term “migralepsy” (migraine followed by seizure) has been revised (Belcastro et al., 2011). Resulting seizures are mostly BTCS (50%), focal seizures with impaired awareness (16%), or focal seizures with unimpaired awareness (8%) (Verrotti et al., 2011). There is no evidence of such comorbidity for migraines without aura.
Cerebrovascular disease
Considering that negative symptoms are invariably associated with cerebrovascular disease, postictal paresis and aphasia could easily be confused with a transient ischaemic attack (TIA) or stroke, and vice versa. In fact, one single-centre study (Amort et al., 2011) showed that one in five TIAs is a mimic, with ES and migraine being the most frequent diagnoses. In patients with unilateral paresis, TIAs were more likely than mimics, and other clinical symptoms including aphasia, dysarthria or sensory loss were not distinguishing features. Memory loss is more indicative of a mimic, particular ES (Amort et al., 2011). Other negative ictal signs, such as ictal blindness, could be easily misinterpreted as a TIA (Manford, 2001).
Narcolepsy/cataplexy
Narcolepsy type 1 (NT1) is a chronic central disorder of hypersomnolence provoked by a loss of hypocretin-producing cells in the posterior hypothalamus (Bassetti et al., 2019). NT1 often arises during childhood, although a correct diagnosis is frequently only made many years after the onset of symptomatology (Luca et al., 2013) with a high misdiagnosis rate (Plazzi et al., 2011). The two main NT1 symptoms are excessive daytime sleepiness and cataplexy. The latter is pathognomonic for the disease and characterized by episodes of muscle weakness, occurring during wakefulness and evoked by emotions. Other manifestations are sleep paralysis and hypnagogic/hypnopompic hallucinations. Cataplexy can involve the whole body resulting in falls, or only limited body areas (e.g. the face). Attacks last from a few seconds to minutes; closely repeated episodes may occur. These manifestations can be misdiagnosed as epileptic seizures, however, the presence of preserved consciousness and an emotional trigger (when evident) is helpful for the differential diagnosis (video sequence 1). It should be noted that interictal and ictal EEG during cataplectic attacks may be characterized by hypersynchronous paroxysmal theta activity (an REM sleep-related phenomenon), which can be misinterpreted as epileptic activity (Vassalli et al., 2013; Baiardi et al., 2015).
Movement disorders
Dystonia is characterized by sustained or intermittent muscle contractions causing abnormal repetitive movements, postures or both. The type of dystonia most often confused with ES is paroxysmal dystonia. The latter occurs as sudden onset dystonia with a self-limiting character. It is triggered by activity or action and lasts as long as the trigger is present (Albanese et al., 2013).
Non rapid eye movement parasomnias
Non rapid eye movement (NREM) parasomnias occur in childhood, adolescents and less commonly in adults. Phenotypes include sleepwalking, sleep terrors and confusional arousals. Further details are outlined in the “childhood” section addressing “disorders of arousal”.
Childhood and infancy
Shuddering / shivering attacks
Shuddering or shivering attacks are benign non-epileptic events that typically begin in infancy. The clinical events consist of rapid shivering of the head, shoulder, and occasionally the trunk. Events are usually brief, lasting not more than a few seconds, with a frequency of up to more than 100 per day (video sequence 2). Although in most cases diagnosis is clinical, a polygraphic video-EEG recording might be necessary to confirm the non-epileptic nature of these events as they can mimic myoclonic, tonic, atonic seizures or epileptic spasms. The pathophysiology of shuddering attacks remains unknown. Parents should be reassured of the self-limiting character of the manifestations, and non-specific measures are needed.
Sandifer syndrome
Sandifer syndrome is a rare complication of gastroesophageal reflux disease, first described more than 50 years ago (Kinsbourne and Warrington, 1964). Clinical manifestations consist of episodes of spasmodic torsional dystonia with arching of the back and opisthotonic posturing and/or spastic torticollis. It is supposed that the positioning provides relief from pain and discomfort caused by the acid reflux, but the pathophysiology is unknown. Because of similarities between the manifestations and epileptic seizures, such as tonic seizures, the patient may undergo lengthy, expensive, and unnecessary neurological testing. This may lead to delay in correct diagnosis and appropriate treatment, or even to the use of inappropriate medication.
Breath-holding spells
Breath-holding spells (BHS) associated with loss of consciousness occur in 0.1 to 4.6% of children, between the ages of six months and five years (Leung et al., 2019). In nearly 20% to 30% of parents of children with BHS, there is a positive family history of similar episodes. The aetiopathogenesis of BHS is thought to be multifactorial, including dysregulation of the autonomic nervous system, vagally-mediated cardiac inhibition, delay in myelination of the brain stem, and iron deficiency-related anaemia. There are two different BHS phenotypes: cyanotic and pallid types. Generally, children present with only one phenotype, but a small minority can experience both. The cyanotic phenotype, the most common, is generally triggered by stimuli such as anger, fear, irritation and frustration (Benbadis, 2009). Moreover, laughing may also represent a trigger (Maayan et al., 2015). Episodes are generally characterized by a short cry followed by apnoea (breath-holding in forced expiration). This results in clinical cyanosis (red or blue-purple skin, especially around the lips) and loss of muscular tone and consciousness. Spells may last a few seconds with the infant recovering quickly with a long-awaited inspiration and resolution of the cyanosis.
Pallid BHS are principally triggered by pain or fear which may induce a marked vagal activation favouring transient asystole. Crying may be absent or minimal (the skin may be pale and sweaty). The length of the apnoea is shorter than that in the cyanotic type, although the evolution of the manifestation is similar (except for an absence of cyanosis).
Longer episodes, in both cyanotic and pallid BHS, may be characterized by decerebrate posturing, trembling movements of the arms and upward eye deviation (a brain stem release phenomenon). The latter may be misdiagnosed as an epileptic manifestation although the whole sequence of abnormal movements is faster. In rare cases, the syncope may trigger a true epileptic seizure (Stephenson, 2012). Generally, based on the settings in which the attacks occur, the precipitating factors, and the temporal sequence of the events, it is sufficient to make a definitive diagnosis of BHS. Finally, EEG recordings do not show epileptic discharges but a slowing and successive flattening of background activity. For both types of BHS, parents should be reassured that the episodes are not dangerous for the child and usually disappear spontaneously within five years of age. However, in children with frequent and prolonged episodes, the ECG should be studied in order to evaluate the risk of life-threatening events (Tomoum et al., 2018). When present, the treatment of anaemia may be helpful (Tatli and Guler, 2017).
Spasmus nutans
Spasmus nutans (SN) is a self-limiting benign clinical entity occurring in early childhood. It is characterized by ocular oscillations, nystagmus, head nodding/bobbing and anomalous head positions (the latter probably represent a compensatory mechanism) (Gottlob et al., 1992). A subclinical nystagmus may persist until the age of about 12 years (Gottlob et al., 1995). SN may be associated with or mimics an underlying retinal pathology (Gottlob et al., 1995). Historically, an association between SN and optic pathway gliomas has been reported. However, these findings have not been confirmed by recent studies (Bowen et al., 2017). SN does not need treatment (if only SN is present). The differential diagnosis with epileptic phenomena is simple, given the persistence and variability of manifestations.
Non-epileptic staring spells
Non-epileptic staring spells are characterized by short episodes (less than 60 seconds) in which the infant is staring, unresponsive with no motor manifestations (Benbadis, 2009). Episodes may have an abrupt onset and cessation. Besides the normal EEG pattern during these spells, the difference with epileptic phenomena is based on the maintained reactivity to touch and absence of playing interruption during the episodes. Moreover, manifestations are first noticed by a teacher or a physician (Rosenow et al., 1998).
Stereotypies
Stereotypies, also defined as mannerisms, are recurring repetitive movements, utterances or postures usually involving the upper extremities (video sequence 2). They can be stopped voluntarily or end when the child focusses attention on a new activity or when he/she is distracted (Stephenson, 2012). Stereotypies are common in patients with intellectual disability and autism spectrum disorder although they can be seen in otherwise healthy children. Clapping or shaking arms are frequently reported. Trigger events are not necessary to elicit stereotypes although periods of stress intensify the frequency. Stereotypes are generally easily distinguishable from epileptic automatisms due to their frequent recurrence during the day, the lack of altered awareness during and after the manifestations, and the absence of other features suggesting a seizure. On the other hand, based on the differential diagnosis of simple stereotypes, tics and other movements such as nodding, the diagnosis may still be challenging (Singer, 2009; Martino and Hedderly, 2019).
Tics
Tics are sudden, rapid and repetitive simple or complex involuntary movements or vocalizations that occur repeatedly during the day. Simple tics may be misdiagnosed as myoclonic epileptic manifestations. Complex tics may present as a coordinated series of movements (purposeful or not) that may resemble focal-onset impaired-awareness seizures. However, both simple and complex tics are characterized by a premonitory urge or subjective relief, following movement that easily distinguishes them from seizures. Moreover, similar to stereotypes, tics may be interrupted voluntarily although generally only for a short period of time. However, the differential diagnosis is rarely challenging, especially in children with intellectual disability or communication problems. In these cases, EEG recordings may confirm the absence of epileptic abnormalities during the manifestations (Bye et al., 2000).
Benign paroxysmal tonic upward gaze
Benign tonic upward gaze (BTU) is a paroxysmal neurological disorder that generally begins in the first year of life, characterized by persistent or occasional sustained conjugate upward eye deviation with flexion of the neck (Ouvrier and Billson, 1988; Humbertclaude et al., 2018). These movements may be followed by downward saccades in order to counteract the upward movement of the gaze. Horizontal movements are preserved. During the manifestations, the child may retain normal eye contact in the superior visual field. The frequency of the attacks may be highly variable, from a few episodes per year to pluri-daily manifestations. Also, the duration is notably variable (from a few seconds to minutes or more). The disorder is often associated with ataxic symptoms. Episodes are generally relieved by sleep and exacerbated by fever and stress. Although BTU is considered a benign disorder, because of negative investigations and frequent spontaneous remission of symptoms over time, children with BTU are more likely to have an abnormal neurological examination and cognitive dysfunction. Moreover, BTU may be associated with structural brain alterations, neurotransmitter disorders, channelopathies and epileptic manifestations (Luat et al., 2007; Blumkin et al., 2015; Humbertclaude et al., 2018).
Benign paroxysmal torticollis
Benign paroxysmal torticollis (BPT) is characterized by recurring episodes of head tilt or flexion of the head on one side. Onset occurs within the first year of life (often in the first three months). The duration of the episodes varies from patient to patient, but usually persists for a few hours or days. Co-occurrence of BPT, BTU and benign paroxysmal vertigo (BPV) has been reported in the same patient or in subjects within the same family (Roubertie et al., 2008; Humbertclaude et al., 2018). The differential diagnosis includes epilepsy, Sandifer's syndrome and posterior fossa tumours. The frequency and duration of the manifestations decrease with age, usually disappearing after the age of five. However, as for BTU, children with BPT have an increased risk of neurological and cognitive alterations (Humbertclaude et al., 2018).
Benign paroxysmal vertigo
Benign paroxysmal vertigo is characterized by a sudden fear of falling, associated with loss of balance and possible falling to the ground. The onset is abrupt and the child is frightened and seeks support, to cling to something. Consciousness is preserved. Nystagmus and neurovegetative symptoms, such as paleness, nausea, sweating and vomiting, may be present as well as abnormal head postures. Episodes generally last less than 5 minutes (rarely more than 24 hours). The frequency of attacks varies, from daily to monthly episodes, and decreases with age. Episodes may be triggered by stimulation such as rides and swings but also by fatigue, stressful events or increases in temperature (Drigo et al., 2001). BPV is considered a childhood migraine variant with age at onset of between two and four years.
Self-gratification
Self-gratification or self-stimulation may occur during infancy and more frequently in pre-school girls. Typically, manifestations consist of pressing, stretching and adducting the legs while sitting or lying, with rhythmic flexion of the hip (video sequence 3). The respiratory rate increases and the child may assume a distant expression with a flushed face. Episodes generally last a few minutes and can be interrupted by intervention. Brief manifestations may be confused with focal seizures with impaired awareness. However, their occurrence only under specific conditions (in a car seat or lying down) is strongly suggestive of non-epileptic manifestations.
Alternating hemiplegia of childhood
Alternating hemiplegia of childhood is a rare neurodevelopmental disorder characterized by recurrent episodes of loss of muscular tone affecting one side of the body (hemiplegia) (Andermann et al., 1995). The condition is considered autosomal dominant, and mutations in the ATP1A3 gene are primarily involved. In some cases, mutations in ATP1A2, CACNA1A, ADCY5, TANGO2 and SLC1A3 genes have been reported. Hemiplegic episodes start in the first few months of life and can be accompanied by other paroxysmal manifestations, such as lateral eye and head deviation towards the hemiplegic side, as well as a peculiar monocular nystagmus (video sequence 4), which may precede the attack. The hemiplegia may alternate from one side of the body to the other, or affect both sides at the same time with possible severe clinical impairment and difficulties in swallowing and breathing (video sequence 4). A unilateral attack, in particular, when accompanied by eye and head deviation, may be confused with focal motor seizures. Hemiplegic attacks may be triggered by various stimuli, i.e. exposure to heat or cold, emotional stress, fatigue, excessive light as well as sound stimuli, trauma, and bathing. The frequency of attacks is high, usually several per month or per week, generally decreasing with age. The duration of an episode is variable, lasting from a few minutes to several hours. A characteristic feature is that all symptoms disappear in sleep and can reappear shortly after awakening. Cognitive delay to a variable degree is a common feature. Epilepsy has been reported in 50% of cases.
Disorders of arousal
Disorders of arousal (DOA) are parasomnias related to NREM sleep, characterized by complex, apparently purposeful, goal-directed behaviours (Derry et al., 2009). They include confusional arousals, sleepwalking and sleep terrors and are identified based on the length of manifestation, the presence of deambulatory behaviours, and association with autonomic and affective (fear) symptoms. DOA are very frequent during childhood. The differential diagnosis between DOA and epileptic seizures, particularly those occurring in patients with sleep-related hypermotor epilepsy (Derry et al., 2009; Proserpio et al., 2019), may be challenging. The prolonged duration of DOA, with a waxing and waning pattern of episodes, the presence of verbal interaction during manifestations, failure to fully arouse the child after events, and their non-stereotypic features strongly support the diagnosis (Derry et al., 2009). Home-video recordings of multiple events can be useful for the diagnosis (Nobili, 2009), however, when episodes are of short duration, video-polysomnographic recordings may be necessary (Derry et al., 2009; Proserpio et al., 2019). Examples of clinical manifestations of some different types of DOAs are shown in video sequence 5.
Migraine, narcolepsy/cataplexy, and psychogenic non-epileptic seizures
The topic of migraine, narcolepsy/cataplexy and psychogenic non-epileptic seizures has already been addressed in the section Adults and adolescents.
Concerning narcolepsy/cataplexy, it should be noted that children have a distinctive phenotype compared to adults (Plazzi et al., 2011). In particular, cataplexy may be misdiagnosed or overlooked, as it may manifest as a movement disorder characterized by alternation of hypotonia and hyperactive movements, also in the absence of emotional triggers (Plazzi et al., 2011; Pizza et al., 2013).
Regarding PNES, a higher frequency of non-motor manifestations (staring, daydreaming) is observed in childhood with respect to adults (Szabõ et al., 2012). Co-occurrence rates of epilepsy and PNES (Kutluay et al., 2010) are equally high in children compared to adults and adolescents, therefore video-EEG recordings may also be required in this age group in order to resolve the differential diagnosis (video sequence 6).
Neonates
In the neonatal period, a variety of motor phenomena can occur that are non-epileptic in nature. Benign neonatal sleep myoclonus and jitteriness are frequent, while other abnormal movements, including neonatal hyperekplexia, are less commonly seen. Most of these phenomena are benign and have no impact on the neonate's neurodevelopmental outcome. However, some phenomena, such as jitteriness could be due to a possible pathology that may require specific investigations and treatment.
Benign neonatal sleep myoclonus
Benign neonatal sleep myoclonus was first described by Coulter and Allen (Coulter and Allen, 1982). Clinical manifestations are characterized by repetitive, rhythmic, high-frequency myoclonic jerks concerning a single, two, three or all extremities, that are synchronous or asynchronous and last for seconds or minutes. The myoclonus may appear in all sleep stages and during the transition from sleep to awakening, although its frequency is greatest during quiet sleep (Resnick et al., 1986; Kaddurah and Holmes, 2009) (video sequence 7). Facial myoclonias are rare (Di Capua et al., 1993). Episodes have been reported to be provoked or worsened by procedures such as repetitive sounds, tactile stimuli, or rocking (Alfonso et al., 1995). Incidence is indicated at 0.8-3/1,000 term and near-term babies (Maurer et al., 2010). Manifestations usually start within a few days after birth and disappear spontaneously at the age of four to six months. The pathophysiology is unknown, but thought to be related to immature myelination of descending inhibitory pathways during sleep and genetic predisposition. Treatment is not unnecessary, moreover, AEDs increase the intensity, frequency and duration of the myoclonus. EEG or video monitoring should be reserved for children in whom the diagnosis is not clear from clinical history, clinical examination and home video. Differential diagnosis with myoclonic epileptic seizures and focal clonic seizures is important, but usually not problematic. Although EEG is normal, central and vertex sharp waves can be observed. Polygraphic analysis shows that these follow and do not precede the myoclonic jerks, and correspond to evoked potentials that are possibly misleading and may be considered as epileptic (Losito et al., 2017).
Jitteriness
Jittery is a term used to describe a series of recurrent tremors in infants (video sequence 8). Tremors are defined as involuntary, rhythmic, oscillatory movements of equal amplitude around a fixed axis, referred to as “fine” when frequency is high (> 6 Hz) and amplitude is low (lower than 3 cm) or “coarse” when frequency is low (< 6 Hz) and amplitude is high (higher than 3 cm) (Painter et al., 1995).
Jitteriness occurring in the neonatal period is observed in about two thirds of healthy newborns, generally triggered when startled, crying or upset, and typically stopped by passive flexion or gentle restraint of the extremity (Armentrout and Caple, 2001). Eye movements or autonomic changes (hypertension, apnoea) do not accompany jitteriness, and their presence could rather suggest their epileptic nature. The differential diagnosis includes myoclonic or clonic seizures; epileptic jerks of clonic seizures tend to be slower at 2-3 per second in contrast to tremors which occur at a faster frequency of 5-6 per second. The pathophysiology of jitteriness is unknown. The condition may be benign or related to pathological conditions such as hypoglycaemia, hypocalcaemia, sepsis, hypoxic ischaemic encephalopathy, intracranial haemorrhage, hypothermia, hyperthyroid state or drug withdrawal.
Hyperekplexia
Hyperekplexia, also known as startle disease, is a rare autosomal, dominantly inherited disorder, characterized by sudden, exaggerated startle responses to unexpected auditory, tactile, visual and vestibular stimuli or strong emotions, and generalized muscle stiffness (video sequence 9). The disease can present in the neonatal period and diagnosis is based on clinical findings, typically by the sudden startle response triggered by tactile stimulation on the glabella. Long-lasting generalized contraction involving the whole body (extremities, trunk and face) may be observed (stiff-baby syndrome), and possible laryngospasm and cardiorespiratory failure increase the risk of sudden infant death. Differential diagnoses are epileptic seizures, tonic seizures when generalized tonic contractions are observed, or myoclonic seizures, as spontaneous myoclonic jerks that are isolated or occur in brief bursts may be observed (Eisermann et al., 2014). Also, neonatal tetany or phenothiazine intoxication should be considered. Hyperekplexia is caused by disturbances in inhibitory glycine-mediated neurotransmission with mutations described in genes encoding glycine receptor subunits or associated proteins, such as (most frequently) the glycine receptor alpha-1 subunit gene (GLRA1) in the brain and spinal nerves, as well as SLC6A5, GLRB, GPHN, and ARHGEF9 (Schaefer et al., 2013). Episodes gradually subside during the first months of life. Patients benefit from low-dose benzodiazepine, valproic acid, and levetiracetam. In cases of prolonged stiffness, a simple postural manoeuvre consisting of gentle flexion of the head and trunk is known to be lifesaving (the “Vigevano manoeuvre”; see video in Eisermann et al. [2014]).
Conclusion
Accurate diagnosis of paroxysmal events can be challenging. No single clinical feature indicates whether a seizure is of epileptic nature or not. Given that there are many differential diagnoses, knowledge of the key features of each paroxysmal event may help to identify patterns of symptoms leading to a clinical diagnosis. Knowledge of the items to aid in identifying the respective aetiology will improve history taking. Patient and witness accounts may be imprecise or events might not have been witnessed. Hence, other diagnostic tests including EEG and MRI will contribute to the final diagnosis. Video-EEG monitoring is the gold standard for diagnosis and should be considered early when attempting to identify the exact nature of paroxysmal events, or when there is doubt or therapy is not effective. Knowledge of clinical features remains a key clinical competency for the diagnosis of epileptic and non-epileptic seizure, for any age group.
Key points:
- •Clinical assessment and diagnosis of paroxysmal events are important core competencies
- •Usually, no core symptoms of paroxysmal events are present during consultation
- •Patients and witness accounts have limited reliability
- •The rate of misdiagnosis of epilepsy and non-epileptic events is high
- •Knowledge of clinical features and differences of various paroxysmal events is important for history taking
- •PNES may have stereotypic semiology
- •Syncope is often misinterpreted as seizure due to convulsions
- •There is no single semiological feature that distinguishes between epileptic seizures and non-epileptic events
- •Videos are a valuable tool in diagnosing paroxysmal events
- •The differential diagnosis of paroxysmal events is specific to different age groups.
Learning points :
- •Benign neonatal sleep myoclonus is a benign non-epileptic condition.
- •The diagnosis is generally based on clinical context and no further investigations are needed.
- •EEG may reveal sharp transients in central and vertex regions that do not precede but follow the myoclonic jerk, and correspond, probably, to sensory evoked potentials. These might lead to misdiagnosis and to unnecessary and ultimately detrimental effects on the infant.
- •The benign nature of this condition should be explained to parents to allay any anxiety about this condition.
- •The neurological outcome is normal.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Disclosures
Dr. LaFrance reports book royalties from Cambridge University Press, and from Oxford University Press, outside the submitted work. Dr. Nobili reports personal fees from EISAI, other from FIDIA Pharma, personal fees from Bioprojet, outside the submitted work. Dr. von Oertzen reports grants, personal fees and non-financial support from Novartis Phama, personal fees from Roche Pharma, personal fees from Biogen Idec Austria, personal fees from Liva Nova, grants from Grossegger & Drbal GmbH, grants from Merck, personal fees from Indivior Austria GmbH, personal fees and non-financial support from gtec GmbH Austria, personal fees and non-financial support from Boehringer-Ingelheim, personal fees from Philips , personal fees and non-financial support from UCB Pharma, personal fees from Almirall , personal fees from Eisai, outside the submitted work; and he is webeditor in chief of the European Academy of Neurology (EAN), co-chair of the EAN scientific panel for epilepsy, and vize-president of the Österreichische Gesellschaft für Epileptologie (Austrian ILAE chapter). Dr. Leibetseder and Dr. Eisermann have no conflict of interest to declare.
Case 1
Case report of adult with psychogenic non-epileptic seizures
W. Curt LaFrance Jr.
Ms. X is a 69-year-old, widowed white female who experienced fugue states in her 40s, finding herself in strange places. A neurological work-up revealed a silent thalamic stroke seen on MRI, and she was managed on valproate with cessation of the events. She was treated as an inpatient a couple of years after the onset of the seizures for a depressive episode when her husband suddenly died of a heart attack. She had resolution of her depression, and she worked and raised her five children.
After experiencing a myocardial infarction, in cardiac rehab, she developed head and hand shaking on valproate, so she was switched to topiramate. In 2003, she found herself in the rescue squad car having had a seizure. Zonisamide was added to her regimen. With a dislocated shoulder, she felt that she had lost her independence, and she recalls feeling “down in the dumps, I woke up and it was like the end of the world”. Later that year, she was hospitalized for nine days for continuous seizures, with her daughter reporting up to 16 seizures per day.
Her neurologist described brief episodes of generalized tonic-clonic activity, with one beginning in the right hand. She was treated with lorazepam, and she was discharged. She had repeated visits to the emergency department, and because of her neurologist's suspicion of non-epileptic seizures (NES), he arranged for an ambulatory EEG. Her son sent her for a psychiatric evaluation, and she was started on sertraline by her internist and was admitted for a diagnosis of major depression. She was discharged to the partial psychiatric hospital program in late summer, during which time she disclosed, for the first time, intrusive memories of being sexually assaulted, which caused a worsening of the depression over eight months. She described worsening of her flashbacks during the video-EEG (vEEG) monitoring, recalling memories of the assault. She had never told anyone of the rape because of her sense of shame.
Her six-hour vEEG captured one clinical event revealing rhythmic, but irregular movements of the legs, alternating left and right, with knee bending, pelvic thrusting, and horizontal movements of the head, and occasional arm movements were noted. This was followed by her ripping the bedrail mats off the bed, stating she wanted to leave, and crying intermittently. No epileptiform abnormality was associated on the EEG just prior to, during, or after the movements. High-amplitude movement artifact was seen in the bilateral posterior regions intermittently during the event. Interictal tracing revealed occasional, brief, less than five-second runs of slightly higher amplitude, mild slowing in the left temporal region, and independently in the right temporal region.
She had no family history of seizures. Her older sister had depression, and her brother died in his 80s with Alzheimer's dementia.
Her examination was significant for slow quiet speech, whilst relaying her history with her hand placed over her mouth. Regarding her mood, she stated “I had a bad night,” and her affect ranged from smiling to tearfulness. She noted passive thoughts of death. Cognitively, she was alert, and scored 23/30 on the Folstein mini-mental status examination, with deficits in the date, and 0 out of 3 after 5-minute recall. She had trouble with serial 7s after one calculation, and spelling the word “world” in reverse. Clock drawing was intact, and she was of average intelligence.
Because of her initial insight into psychological processes manifesting as neurological signs, and her desire to learn more about the association, psychotherapy was recommended to address underlying issues and comorbidities of NES. She used a seizure diary to document the frequency, precipitants, and description of her seizures. She is being followed by a psychiatrist, who is seeing her for medication management. At this point, she is ambivalent about pursuing a course of psychotherapy.
Examples of videos of PNES can be found in the online supplementary material of the textbook: LaFrance Jr WC, Schachter SC. Gates and Rowan's Nonepileptic Seizures. 4thE. New York: Cambridge University Press; 2018.
Supplementary video material is available at: www.cambridge.org/core/access-codes
Case 2
Monika Eisermann
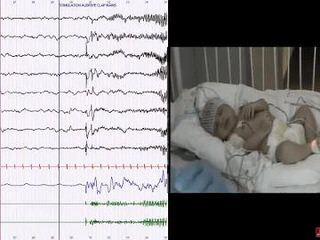
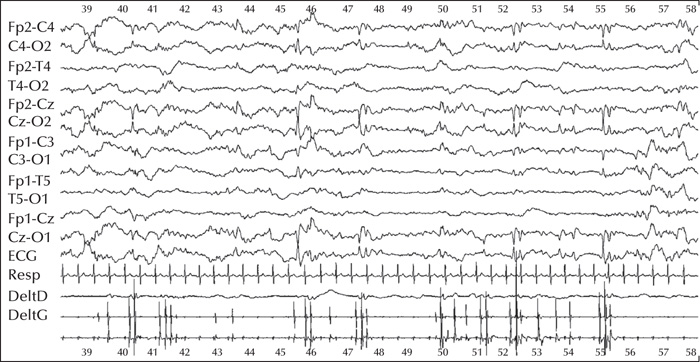
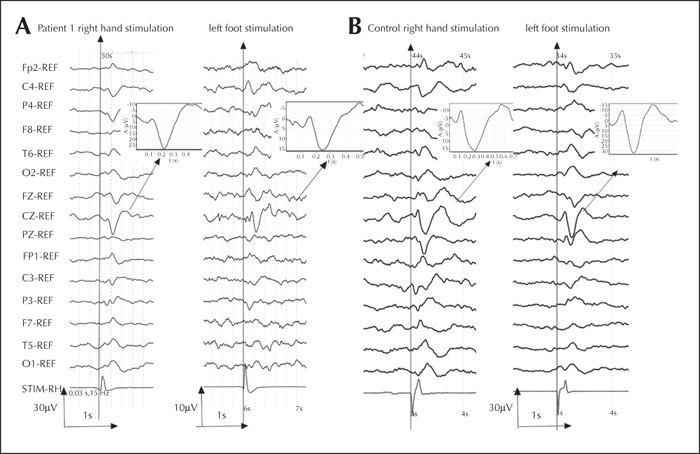
A 16-day-old term baby was transferred from primary maternity to a tertiary referral center with a diagnosis of early-onset myoclonic epilepsy, refractory to triple therapy (phenobarbital, phenytoin, and keppra). Pregnancy and delivery were uneventful. Clinical examination was normal, apart from abundant myoclonic jerks affecting the four limbs, unilaterally or bilaterally, mostly in brief runs, occurring exclusively in quiet sleep. An EEG was performed on Day 16 during quiet sleep (figure 1).
How do you interpret this EEG?
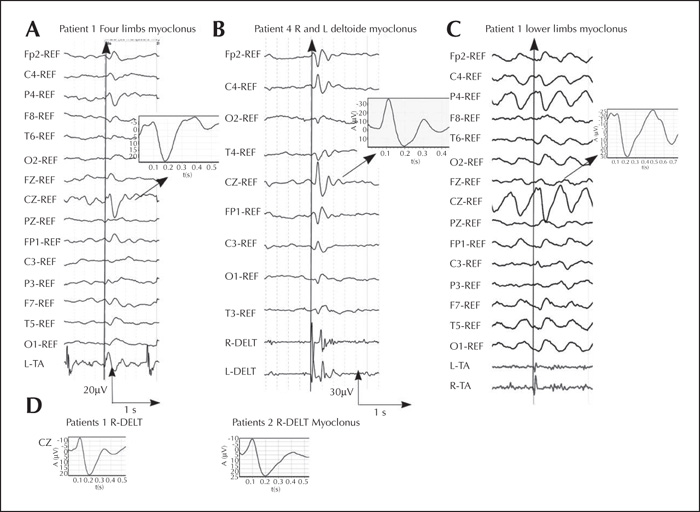
Answer: EEG displays sharp transients in central and vertex regions following (NOT preceding) the myoclonic jerks recorded on surface EMG leads, corresponding to somatosensory evoked potentials (and not to epileptic activity).
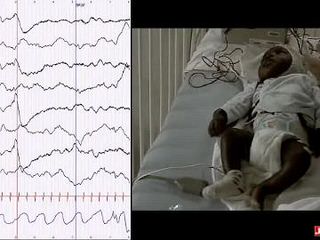
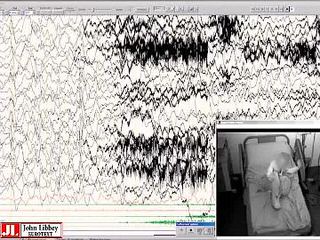
Back-averaging of myoclonic jerks are shown in figure 2, and back-averaging of somatosensory evoked potentials by foot and hand tapping in figure 3.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License