Epileptic Disorders
MENUEarly-onset epileptic encephalopathy with migrating focal seizures associated with a FARS2 homozygous missence variant Volume 22, issue 3, June 2020


- Key words: epilepsy of infancy with migrating focal seizures (EIMFS), FARS2, aminoacyl tRNA transferase, lactate
- DOI : 10.1684/epd.2020.1168
- Page(s) : 327-35
- Published in: 2020
Epilepsy of infancy with migrating focal seizures (EIMFS) is now a well-recognized early-onset syndrome included in the ILAE classification of the epilepsies. KCNT1 gain-of-function variants are identified in about half of patients. In the remaining cases, the underlying genetic component is far more heterogeneous with sporadic mutations occasionally reported in SCN1A, SCN2A, SLC12A5, TBC1D24, PLCB1, SLC25A22, and KCNQ2.
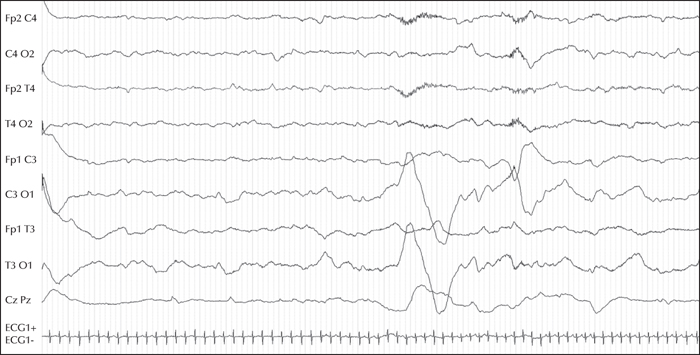
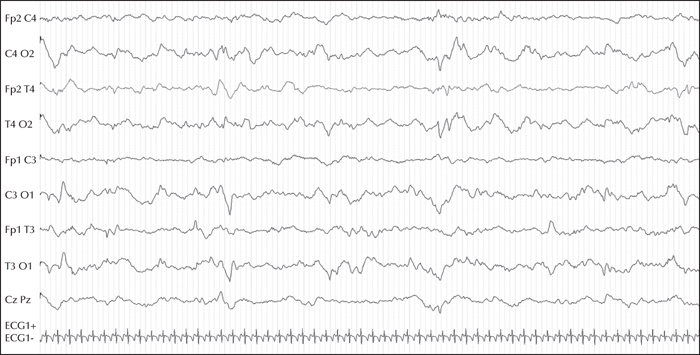
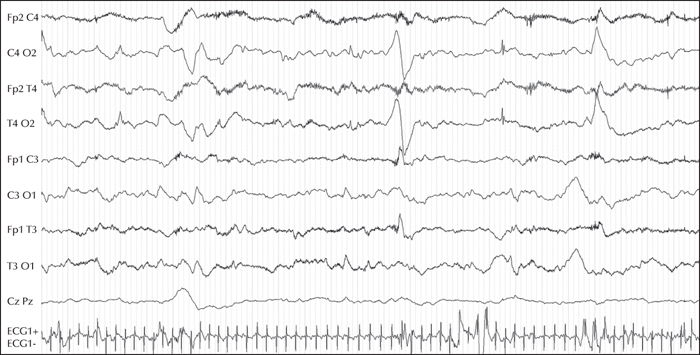
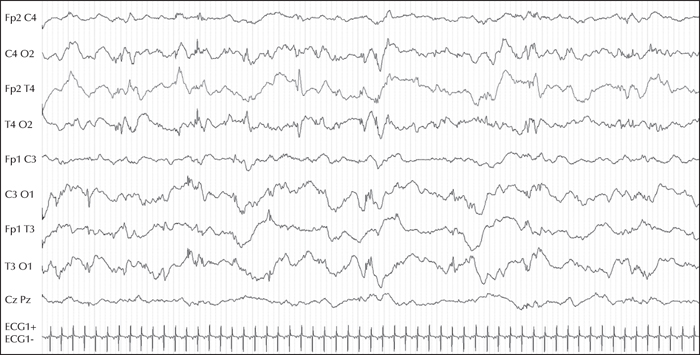
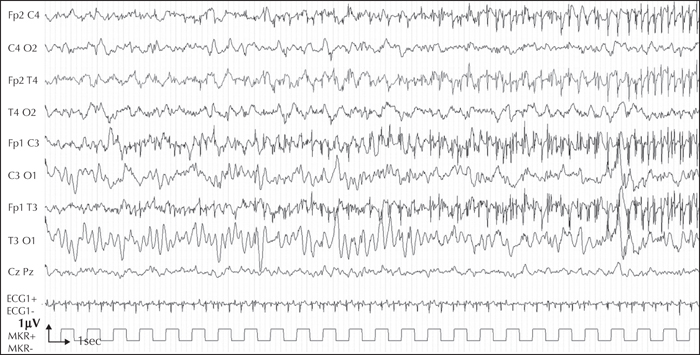
Here, we report, for the first time, a homozygous deleterious variant in the FARS2 gene, identified using a 115-gene panel for monogenic epilepsies, in a patient with EIMFS. This boy was the second child born to healthy consanguineous parents. The first seizures occurred at six weeks of age. The patient rapidly developed severe epilepsy with focal discharges on EEG, migrating from one brain region to another, highly suggestive of EIMFS. At five months of age, he had daily multifocal clonic seizures and erratic myoclonic fits, which were not consistently related to spikes or spike-and-wave discharges. Neurological status was severely abnormal from onset and the patient died at 10 months of age from respiratory distress. Using the gene panel, a homozygous missense variant of FARS2 was identified,at Chr6 (GRCh37):g.5404829C>T, c.667C>T (NM_001318872.1), inherited from both parents, leading to an arginine-to-cysteine substitution, p.(Arg223Cys).
FARS2 is a member of the mitochondrial aminoacyl tRNA transferase (ARS) enzymes. ARS variants are increasingly recognized causes of early-onset epileptic and neurodevelopmental encephalopathies, however, the associated epileptic phenotype is not completely described. This case shows that FARS2-related seizures can mimic EIMFS in the early stage of the disease. Furthermore, in the setting of migrating focal seizures of infancy, FARS2 should be considered as a further candidate gene, and increased lactate level and occurrence of refractory myoclonic seizures are possible key features to suspect FARS deficiency.