Epileptic Disorders
MENUDevelopmental and epileptic encephalopathies: recognition and approaches to care Volume 23, issue 1, February 2021


Learning objectives for the ILAE curriculum
(1) Understand the definition of a person with a developmental and epileptic encephalopathy.
- A.Be able to expand on the concept of whether neurocognition and behavior are influenced or not by epileptic activity.
- B.Be able to give examples of case illustrative scenarios of DEE and DE.
(2) Be aware of the group of patients who may be most at risk of DEE.
- A.Understand the relevance of identifying those in whom control of epilepsy influences DEE expression with regard to investigations, treatment and prognosis.
- B.Understand the setting where the term DEE is appropriate.
(3) Understand the differences in the profile and management of patients with DEE related to, and not related to, epilepsy.
- A.Describe the common etiologies.
- B.Describe the approach to care for a patient with DEE.
- C.Describe factors which are key to cognitive outcome.
Delineating the underlying etiologies and recognizing electroclinical syndromes for people with epilepsy is critical for best care practice. This text provides an overview of the meaning of developmental and epileptic encephalopathies and it underlines how correctly classifying patients might affect their management and outcome expectations. People with poorly controlled and recurrent seizures are compromised from reaching their full potential. It is important to approach the clinical setting inclusive of all variables which can influence neurocognition and behavior. The assessment of an encephalopathic patient with epilepsy should include acquired and reversible elements, for example, high levels of antiseizure medications (ASMs) as well as newly acquired brain infections [1].
Evolution, definitions and use of the terms “developmental” and “epileptic” encephalopathy [2-6]
The original concept of epileptic encephalopathies (EE) is related to conditions whereby the abundant epileptiform abnormalities and/or high number of epileptic seizures themselves contribute to cognitive regression [7].
In contrast, in conditions where the cognitive development and behavior is impaired independently to the epilepsy onset, and epilepsy is characterized by a high frequency of seizures and abundant epileptiform abnormalities, the term “developmental and epileptic encephalopathy” (DEE) is more appropriate: this was proposed in the 2017 revision of the classification [2, 3]. As such, the term DEE is appropriate to use when both developmental impairment and epileptic activity have impact on the cognitive and behavioral state of the affected person [2]. Most patients with DEE have a genetic etiology, whereby the genetic variant is responsible for both cognitive impairment and severe epilepsy: in such cases, even with control of seizures, the cognitive outcome is expected to be poor.
The term “developmental encephalopathy” (DE) is a separate entity to DEE [4]. The term “developmental encephalopathy” should be used in the setting of a person with developmental delay, or intellectual disability, due to a non-progressive brain state who also has co-existing epilepsy. Of note, the degree of disability may become more evident with brain maturation. The risk of epilepsy in these people is higher than in the general population but not to the extent that the epilepsy itself causes an epileptic encephalopathy.
An epileptic encephalopathy (EE) occurs when the epilepsy and/or the epileptiform activity specifically affects cognitive and behavioral functions. This is typically seen in patients whose preceding level of function was normal or near normal. In such cases, aggressive treatment should be considered and this might improve the outcome [6].
The 2016 discussion on the classification promoted that the terms “developmental encephalopathy” and “epileptic encephalopathy” could be used either independently or together as in DEE. Clinicians are encouraged to select the terms that most aptly apply to the conditions they are dealing with [3]. This was reiterated in the 2017 classification paper which supported the use of the term which best related to the causative etiology for cognitive delay [2]. It is, however, recognized that understanding which is the leading cause between developmental factors and epileptiform activity can be very challenging.
Table 1 summaries the key differentiating features of DE, EE and DEE.
Etiology and epidemiology
Early detection of infants and children who may have DEEs must commence with a high level of suspicion and continuous monitoring. Risk factors to be monitored for are based on the etiologies, clinical seizure semiology, co-morbidities (movements, social interaction change), systemic signs (e.g. neurocutaneous markers) and investigational findings (e.g. EEG, metabolic screens, genetic examinations) [4].
Usually, DEEs manifest during early infantile or childhood periods, although any age group inclusive of adults can be affected [4]. Most patients with DEEs are now recognized to have etiologies due to genetic variants [8, 9]. Prior to this, the de novo or sporadic nature of these genetic variants led to many patient diseases being considered to be due to “idiopathic” or acquired insults. Genetic analysis of a DEE cohort (n = 197) revealed that almost one third had pathogenic variants in known or novel genes [10]. As such, an expanding number of genetic variants are associated with an increased risk of expressing DEE [5].
Other etiologies associated with DEE include structural (e.g. tuberous sclerosis complex (TSC), hypothalamic hamartomas, hemimegalencephaly), metabolic (e.g. pyridoxine or biotinidase deficiency, GLUT1 deficiency) and immune disorders (e.g. Rasmussen syndrome). [4, 6].
Disease expression for different genetic mutations may vary in the subsequent type and severity of epilepsies. For example, KCNQ2 and SCN2A mutations are associated with a range of disease manifestations from mild to severe [11, 12]. Developmental function may vary during different time windows of brain maturation, as an example, children with Dravet syndrome have normal development for the first few years of life but suffer with a plateau in their cognition in early childhood. In addition, the stages of the disease itself will lead to different types and severity of the epilepsies, for example, children with West syndrome develop epileptic spasms typically during infancy, and children with Dravet syndrome suffer various seizure types at different age periods [4]. Understanding these pathways may be very challenging. The neonatal-onset DEEs are illustrative examples, they are often caused by mutations in SCN2A, KCNQ2 and STXBP1, and development may be profoundly impaired and never normal [13]. Seizures may remit during infancy but development will remain severely affected [14].
Ongoing epilepsy and epileptic seizures can result in altered neurogenesis, synaptic reorganization, and disruption of the normal network organization [5]. This results in inhibition of distant brain function connected with the epileptic focus, leading to alteration of neuronal development and transient effects on information processing [6]. In addition, the immature brain can be permanently modified by neuroinflammation which is evident in this setting.
The 2020 overview by Scheffer and Liao lists the following as important examples under the concept of DEE: Ohtahara syndrome, early myoclonic epilepsy, epilepsy of infantile onset with migrating focal seizures, West syndrome, Dravet syndrome, myoclonic-atonic epilepsy, Lennox-Gastaut syndrome, focal lesional epilepsies (e.g. TSC, hemimegalencephaly, hypothalamic hamartoma) and Rasmussen syndrome, but also the epilepsy aphasia spectrum disorders, i.e. Landau-Kleffner syndrome and continuous spike and wave in sleep (CSWS) [4]. The inclusion of the epilepsy aphasia spectrum epilepsies, or epileptic encephalopathy with continuous spike and wave during sleep (EE-CSWS) electroclinical syndromes, under the heading of DEE, reflects the earlier point about the challenges of delineating the key driving force for the encephalopathy. In this later case of EE-CSWS, the epileptiform activity is the key element to the encephalopathy, but the outcome is still driven by the etiology, hence despite efficacy and promptness in the interventions for the epilepsy control, the epilepsy alone is not the only factor influencing cognitive functions [15].
Routine investigations
Beyond collating the clinical history for risk factors and clinical markers, as well as seizure semiology, diagnostic investigations can be very helpful in identifying an electroclinical syndrome and differentiating DEEs from other conditions.
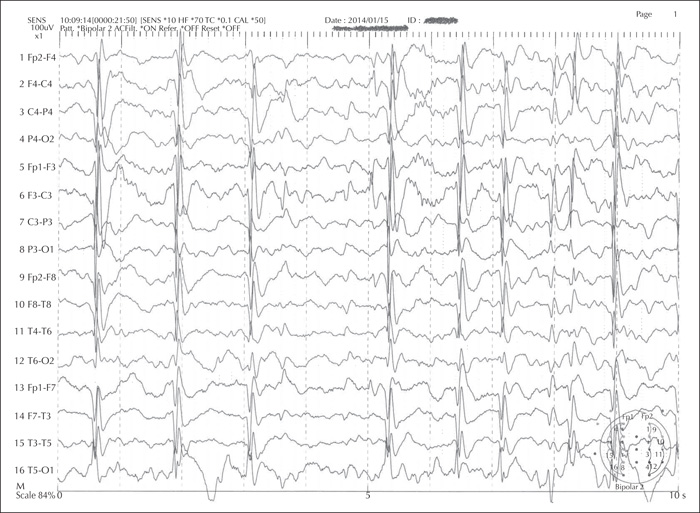
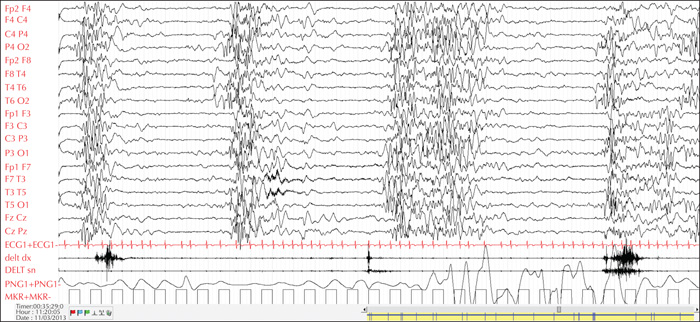
Electroencephalograms (EEGs) can assist in distinguishing different syndromes. Certain electrical patterns are found with specific syndromes such as suppression burst in Ohtahara syndrome, hypsarrhythmia in West syndrome, continuous slow (typically 1.5-2-Hz) spike-and-wave discharges during sleep in CSWS, and spike-wave discharges at a frequency of < 2.5 Hz in Lennox-Gastaut syndrome (LGS) [16] (figures 1, 2). Non-convulsive status epilepticus, known to occur especially in myoclonic-atonic epilepsy, can also be detected on the EEG [17]. In patients with tuberous sclerosis complex (TSC), serial EEGs are recommended in order to detect subtle or sub-clinical seizures, to allow early initiation of treatment [18].
Neuroimaging can be helpful in identifying structural abnormalities, such as malformations of cortical development or hypoxic injuries. Brain MRI is the gold standard recommended for investigation of infants and children with epilepsy [19].
Other investigations would include metabolic workup and immune testing. Certain metabolic disorders, such as biotinidase and pyridoxine deficiency, if detected and treated early, can lead to a favorable outcome [20].
A comprehensive genetic examination with next-generation sequencing (NGS) techniques is suggested in all cases with clinical features suggestive of DEE [8, 21, 22].
Implications for care
In general, for individuals with DE, interventions to control seizures do not change the baseline level of functioning, however, reducing the seizure burden might lead to an improvement in quality of life. Deterioration in seizure control can occur, and during these transient phases, more aggressive seizure management is appropriate. These periods of seizure escalation, which are usually acute, are not associated with change in the level of cognitive functioning. However, these seizures do have impact on the quality of life, as well as the patient's safety, and as such need intervention. An extreme version would be the onset of status epilepticus in the setting of an intercurrent respiratory infection. It would be important to manage the acute seizures and the respiratory infection but to revert to baseline care once the provoking factor is resolved. As such, chronic polypharmacy or high levels of ASMs should be avoided as they are more likely to be detrimental to a person with DE [5].
Patients with EE who develop a specific epilepsy-driven encephalopathy do require aggressive antiseizure interventions as this can lead to significant improvement in cognition and behavior [5, 6, 15]. In this setting, suppression of seizures and epileptiform abnormalities of the EEG might lead to an improvement in cognitive function although not necessarily a return to normal function (figure 3A). Further, the underlying etiology remains key to the long-term outcome [15, 23].
Patients with DEE with cognitive development, based on the dual influences of their developmental baseline and their epileptiform activity, must have their management balanced against realistic expectations for improvement in epilepsy control but not at the cost of adverse effects from over-medication. Uncontrolled seizures and excessive epileptiform abnormalities could worsen the clinical state of a patient with DEE. Whilst attaining control of the typically refractory seizures may be challenging, treatment is still indicated for safety and quality of life. As such, the intervention should be balanced against pragmatic expectations for neurocognitive improvement [6]. Good illustrations of the potential impact of targeted treatment are provided by the gene-related DEE group, which include recommendations for new or repurposed compounds, antisense oligonucleotides, and possibly gene therapy [8].
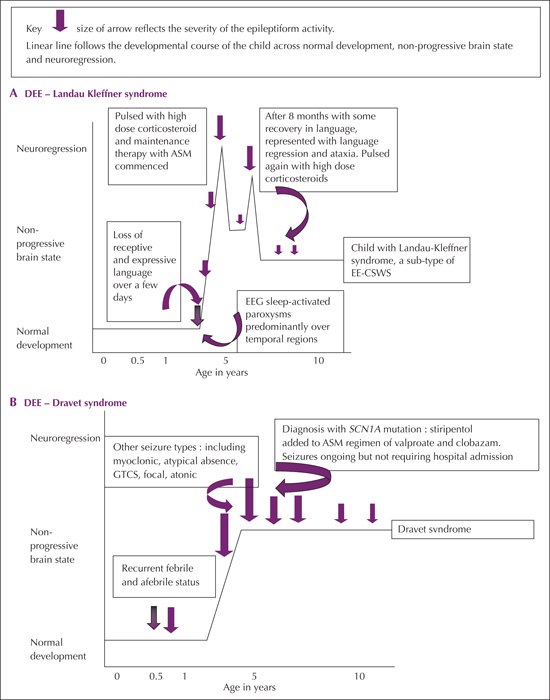
Illustrative scenarios (figures 3A-E)
One of the most well-characterized DEEs is Dravet syndrome(DS), which presents with prolonged febrile, often hemiclonic, seizures within the first year of life in otherwise healthy children, evolving into intractable febrile and afebrile seizures [6, 24]. Dravet syndrome is a good example of DEE as the genetic variant results in direct impact on cognitive function and the child's epilepsy, which manifest at different stages with brain maturation. Development is initially normal in most cases but is delayed with a plateauing in skills, evident generally by two to three years of age. The loss of, and plateauing in skills is felt related to the influence of SCN1A mutation which is the causative gene in 80-85% of affected children [24, 25]. Epilepsy types vary with age, from the prolonged febrile triggered seizures in infancy to afebrile generalized tonic-clonic to myoclonic and atypical absence seizures appearing between the age of one and four years. The specific SCN1A variant influences the overall outcome of the seizures and cognitive function of the affected individual [26]. Figure 3B illustrates the time course of a typical patient with Dravet syndrome, demonstrating how cognitive function changes as the child grows older and how the additional influence of the epilepsy impacts on this.
West syndrome typically presents in the infantile age group, and parents often report subtle regression in the days prior to the clinical events. With the onset of the epileptic spasms (ES), the loss of skills becomes more evident. A race is then in place to initiate intervention, in most cases with corticosteroids [27]. Studies support that as the lag time to treatment extends beyond three to four weeks, the reversal and regaining of previously established skills lessens [28]. In most cases, even with early intervention and resolution of spasms, the underlying etiology will still drive the subsequent cognitive outcome [29, 30]. As such, in-line with the underlying etiology, the infant may have a DEE related to equal influence from the developmental state and the epilepsy, but there are other settings where, following control of the epileptic spasms, return to normal cognitive function occurs and the infant has an EE. Figure 3C (Scenarios 1 and 2) provides illustrations of these two scenarios. Scenario one is an infant who, following a hypoxic birth insult, developed further neuroregression with the onset of ES. The infant then experienced quiescence of seizures and some regain in skills following corticosteroid intervention, but subsequently went on to develop LGS with additional loss of skills before reaching a developmental plateau again. This infant had a DEE. The second infant (Scenario 2) had normal initial development, but suffered with neuroregression, immediately prior to the onset of epileptic spasms. Treatment was initiated within two weeks of seizure onset and was followed by resolution of seizures and return to normal development. Comprehensive structural, metabolic and genetic assessments were unremarkable. This infant had an EE.
Ohtahara syndrome, an early-onset epilepsy syndrome, is a DEE, with drug-resistant tonic seizures evident in the first weeks of life. The EEG is severely abnormal with burst-suppression, and these infants have very poor cognitive outcome. However, the onset is typically too early to assess whether regression occurs, as their pre-symptomatic developmental level cannot be accurately measured. The underlying etiology is often a significant cerebral malformation, e.g. lissencephaly, and as such, these infants can be profoundly delayed regardless of whether they have ongoing seizures [31].
Lennox Gastaut syndrome is a descriptive term for an epilepsy syndrome, always associated with developmental delay and generally diagnosed at around two to five years of age [21]. The EEG usually records slow spike-and-waves and multifocal spikes and there are multiple seizure types; usually tonic but also myoclonic, atypical absence, atonic and tonic-clonic seizures. In these children, the dual impact from the developmental status and the ongoing, often refractory, epilepsy conforms with a DEE. Periods of seizure exacerbation may temporarily result in further loss of skills, e.g. ambulation, but this should return to baseline function as the epilepsy remits or responds to treatment.
An example of DE would be an infant with a non-progressive brain state, which may have occurred due to hypoxia at the time of birth, or the sequelae of meningitis, who, as a result, has global developmental delay and intermittent seizures. Whilst the seizures may be medically refractory or intermittent, there is little change in the functional state of the affected individual. Periods of apparent control or quiescence in the epilepsy do not alter the resting state of the individual.
The causative etiology can manifest differently. A child with tuberous sclerosis complex (TSC) may have neurocognitive and behavioral dysfunction with occasional focal seizures. Despite control of these seizures, it is unlikely that there will be a change in the baseline state of the child, whether the seizures are controlled or not. In this setting, the child would be viewed to have a DE. However, if another patient with TSC and developmental delay develops epileptic spasms, in this setting, the epilepsy would be expected to have a direct and independent impact on the cognition and needs urgent and aggressive antiseizure medication. This child has a DEE. As stated earlier, infants with TSC are recommended by the international guidelines to undergo serial EEGto permit early identification of epileptiform activity. Studies have commenced with pre-clinical seizure intervention with vigabatrin [18]. A prospective study which included a pre-symptomatic arm, at the point at which EEG abnormalities were identified, revealed better cognitive outcomes in the early treatment group, rather than the reactive treatment arm [32, 33]. The EPISTOP study is assessing the impact of initiation of treatment in children with TSC with EEG abnormalities, prior to clinical seizures developing [34]. So, for this case whilst the developmental outcome is influenced by the TSC disease, the early intervention based on the EEG changes further influences the developmental outcome.
A similar setting may occur in a child with trisomy 21, who develops epileptic spasms with regression. In this setting, effective treatment of spasms may lead to improved function and return to baseline. This child is manifesting with a DEE. However, another child with trisomy 21 who has the typical developmental issues and infrequent seizures, which are of minimal impact on the child and controlled with ASMs, would be regarded as having a DE, as illustrated by figure 3D.
Case 1: West Syndrome
Zoe, a six-month-old girl, was referred to the pediatric outpatient department with “abdominal cramps”. Her mother reported that these started when Zoe was four months old. She had taken Zoe to numerous doctors who diagnosed her with a variety of conditions such as colic, gastro-esophageal reflux disease and myoclonic seizures. Her mum was worried because the events were worsening. In addition, Zoe stopped smiling and looking at faces, and “just wasn’t herself”. The events would occur primarily just after she was waking up from a sleep or as she was about to fall off to sleep. She would flex her neck and extend her arms whilst her hips and knees were flexed, lasting a few seconds. This would occur in clusters and Zoe would be very distressed afterwards, even crying sometimes.
Zoe was the first born in her family and had a complicated perinatal course. Her mother had a prolonged labor; Zoe developed fetal distress and was delivered via emergency Caesarian section. She did not cry at birth. Her Apgar scores were 3 and 5 at 1 and 5 minutes, respectively. Zoe was diagnosed with hypoxic ischemic encephalopathy and her management included therapeutic hypothermia. She was admitted to the neonatal unit for a total of three weeks.
She had global developmental delay, however, since these events started, she has lost skills, i.e. regressed.
On examination, Zoe had microcephaly, was not dysmorphic, and did not have any neurocutaneous stigmata. On neurological examination, she was not fixing or following, and had truncal hypotonia with increased tone in her limbs. Reflexes were brisk throughout with spread.
Her EEG showed hypsarrhythmia with intermittent generalized suppression. Zoe was diagnosed with West syndrome and her parents were counselled. Treatment with high-dose corticosteroids and vigabatrin was initiated and her events subsided. MRI of her brain showed delayed myelination for age, thinning of the corpus callosum and generalized brain atrophy. Once events were under control, Zoe started to regain some developmental milestones, returning to her pre-seizure baseline but remaining significantly developmentally delayed for age. Her course is compatible with a developmental and epileptic encephalopathy.
Case 2: Landau-Kleffner epilepsy
Siya, a five-year-old, previously healthy boy was referred to the pediatric neurology service with a three-week history of not following instructions. This was shortly followed by speaking less and less until he was not talking at all. His father felt that he was still able to hear, as he would react to loud noises and would hear sounds such as the doorbell ring, however, he would not go to the front door as he had previously done. There were also times in the past three weeks when his pre-school teacher and parents would notice him staring ahead for a few seconds. During this time, he would not respond when he was called or touched.
Siya did not have any significant medical illnesses in the past, birth history was unremarkable, and all developmental milestones were appropriate for age. His anthropometry, general, neurological and systemic examinations were normal. It was noted that he was very busy and easily distracted. Speech therapy assessment reported that he had a receptive and expressive aphasia.
His EEG showed centro-temporal spike-and-wave discharges and during sleep, the EEG revealed continuous slow spike-and-wave discharges at 1.5-2-Hz, bilaterally. Brain MRI and metabolic workup were normal. The diagnosis of Landau-Kleffner syndrome was made; his parents were counselled and treatment was initiated. Siya's course was compatible with a developmental and epileptic encephalopathy outcome, which is driven predominantly by the control of the epileptic activity.
Relevance
Early recognition of DEE has far reaching implications for counselling, treatment and prognosis [5, 6]. The underlying etiology may directly influence the severity of the epilepsy and the baseline developmental level of functioning, e.g. KCNQ2, STXBP1[35, 36]. The outcome may remain poor even though seizures are controlled. However, targeted therapies can be effective for seizure control in some DEEs such as for Dravet syndrome with use of stiripentol [37]. Further, in the setting of Dravet syndrome, agents known to exacerbate seizures, such as carbamazepine, on withdrawal, lead to functional improvement [5, 38].
As such, confirming the etiology is extremely important to support targeted interventions such as surgery, vitamin replacements, dietary interventions, specific ASMs and avoidance of ASMs associated with seizure exacerbation [4, 23]. Cognitive outcome is not only related to the cause but also to appropriate treatments, such as hormonal therapy or early surgical treatment in cases with focal cortical dysplasia [6].
Conclusion
In cases in which the encephalopathy is caused by both developmental impairment and epileptic activity, a developmental and epileptic encephalopathy should be considered. Despite the inevitable developmental sequelae for many of the DEEs, early recognition and intervention permits optimal and often improved outcomes. Clinicians should suspect the influence of epilepsy if there is developmental delay which correlates with seizure onset and improves with epochs of better control and frequent seizures, and epileptic discharges on EEG. It is helpful to identify etiologies and electroclinical syndromes as this can assist with prioritizing investigations and selecting best treatments. Insight into situations where the child has a DE will ensure that aggressive ASM interventions are avoided. The approach to care should focus on optimal understanding of the disease process and the driving causes of the encephalopathy.
Key points
- •A developmental and epileptic encephalopathy occurs when both developmental impairment and epileptic activity independently influence the neurocognitive and behavioral outcome
- •Many DEEs are related to gene mutations and present in infancy or early childhood.
- •Timeous initiation of treatment for people with DEE can result in improved seizure control, and in addition to precision therapy, may lead to improved cognitive outcomes.
- •Interventions for people with DEE should be based on a balance between control of epileptic activity and avoidance of unacceptable adverse effects.
- •Underlying etiology is a leading aspect to subsequent cognitive outcome.
- •Improved seizure control for people with a predominantly non-progressive brain state (e.g. cerebral palsy) may not necessarily alter the behavioral or cognitive outcomes, and in this setting, aggressive ASM intervention should be avoided.
Disclosures
None of the authors have any conflict of interest to declare.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License