Epileptic Disorders
MENUDescription of a peculiar alternating ictal electroclinical pattern in a young boy with a novel SPATA5 mutation Volume 22, issue 5, October 2020


- Key words: SPATA5, epileptic encephalopathy, SPATA5 syndrome, phenotype
- DOI : 10.1684/epd.2020.1204
- Page(s) : 659-63
- Published in: 2020
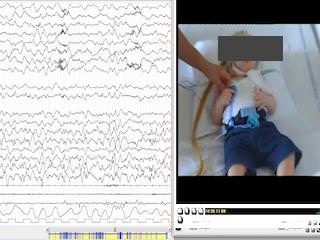
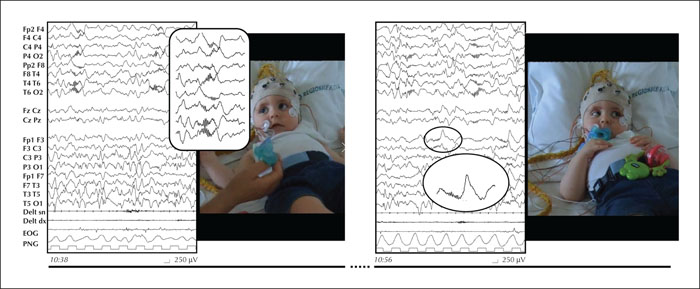
Heterozygous variants in the SPATA5 gene have recently been described to be associated with epileptic encephalopathy. As of 2019, 37 patients have been described in the published literature. We report a patient with a novel autosomal recessive pathogenic variant in SPATA5 and a clinical phenotype consistent with SPATA5 syndrome, including severe neurological impairment, intellectual disability (ID), generalized intractable epilepsy, microcephaly, abnormal muscle tone, and sensorineural hearing loss. The epileptic clinical features were characterized by infantile spasms associated with seizures with a complex ocular movement; a predominant involvement of the posterior cerebral area and cortical visual impairment were also noticed. This phenotype is highlighted with a review of the literature showing other patients with SPATA5-related disease. This report aims to contribute to further understanding phenotype/genotype correlations, which are fundamental for the interpretation of data made available by exome sequencing for the diagnosis of epileptic encephalopathies. [Published with video sequence].