Epileptic Disorders
MENUClinical evolution and epilepsy outcome in three patients with CDKL5-related developmental encephalopathy Volume 21, issue 3, June 2019

Authors
1 Department of Paediatric Neurosciences, Santobono-Pausilipon Children's Hospital, Naples
2 Department of Translational Medical Sciences, Child and Adolescent Neuropsychiatry, University of Naples Federico II, Naples
3 NESMOS Department, Sant’ Andrea Hospital, “Sapienza” University of Rome, Rome
4 Regional Referral Centre for Neurofibromatosis, Department of Woman, Child, General and Specialistic Surgery, “Luigi Vanvitelli” University of Naples, Naples
5 Department of Neuroscience, Reproductive and Odontostomatological Sciences, Epilepsy Centre, Federico II University, Naples
6 Pediatric Neurology and Muscular Diseases Unit, IRCCS Istituto “G. Gaslini”, Genova
7 Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genova, Genova, Italy
* Correspondence: Pia Bernardo
Department of Paediatric Neurosciences,
Neuropsychiatry Unit,
Santobono-Pausilipon Children's Hospital,
Via Mario Fiore n.6, 80129, Naples, Italy
- Key words: CDKL5 gene, epileptic encephalopathy, neurodevelopmental encephalopathy, genetic epilepsy, honeymoon period, developmental delay, Rett syndrome
- DOI : 10.1684/epd.2019.1071
- Page(s) : 271-7
- Published in: 2019
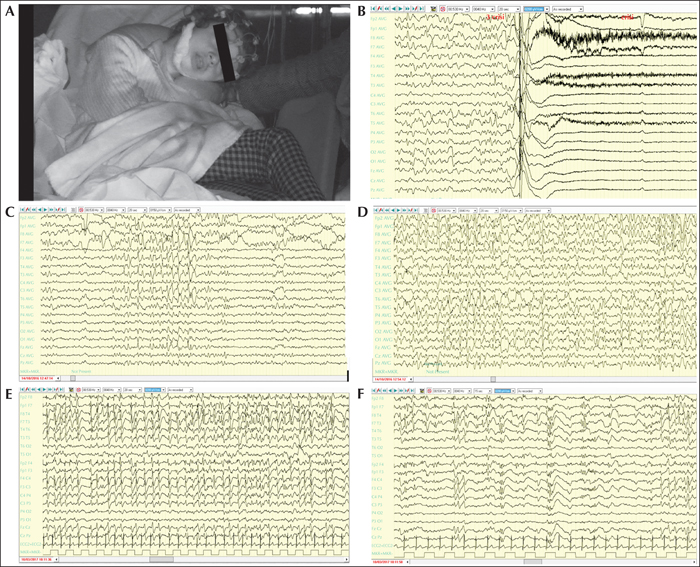
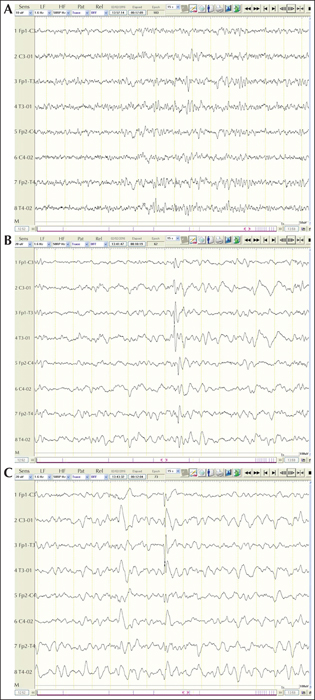
Aims
To further characterise CDKL5-related disorder, previously classified as an early-onset seizure variant of Rett syndrome, which is currently considered a specific and independent early-infantile epileptic encephalopathy.