Hépato-Gastro & Oncologie Digestive
MENUDuodenopancreatic involvement in MEN1 Volume 29, issue 1, January 2022

Figures
Tables
- Key words: MEN1, neuroendocrine tumors, gastrinoma, insulinoma
- DOI : 10.1684/hpg.2021.2301
- Page(s) : 12-23
- Published in: 2022
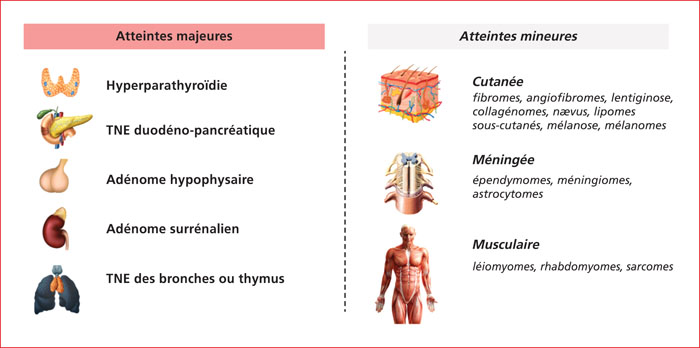
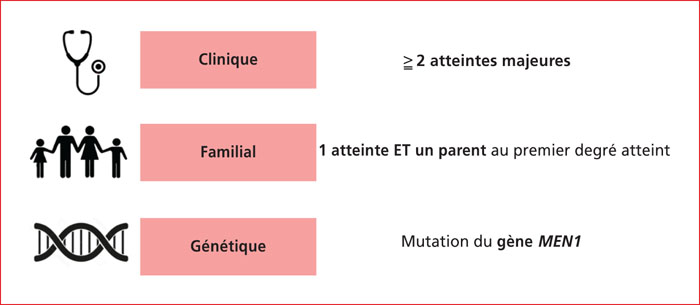
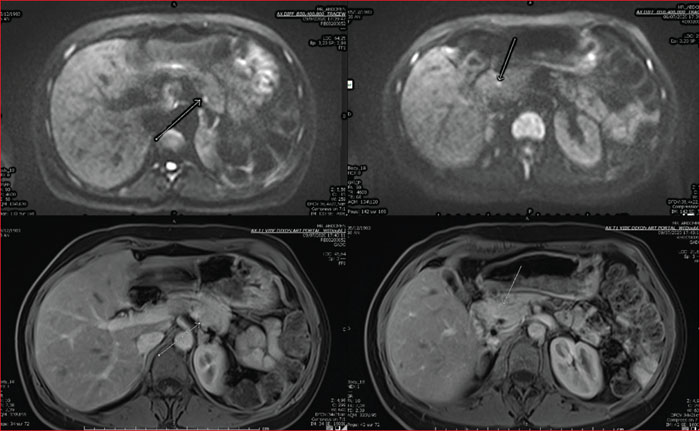
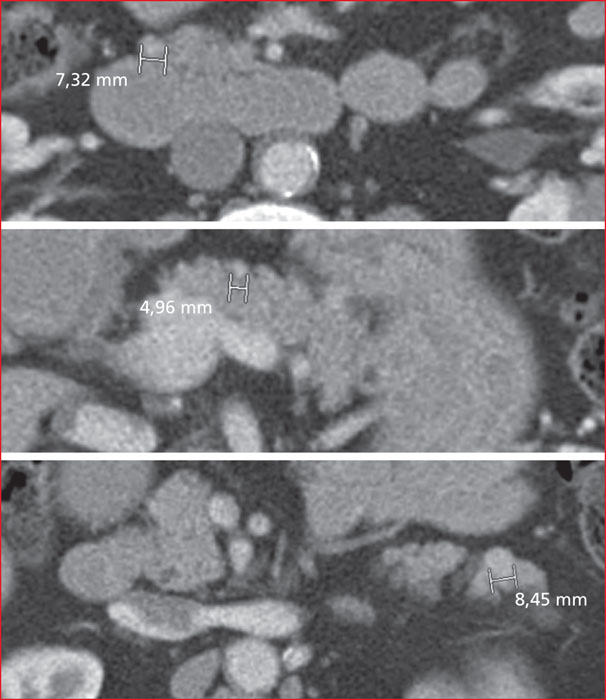
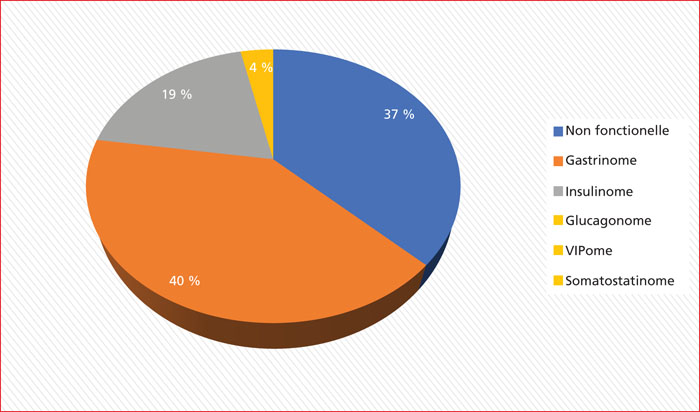
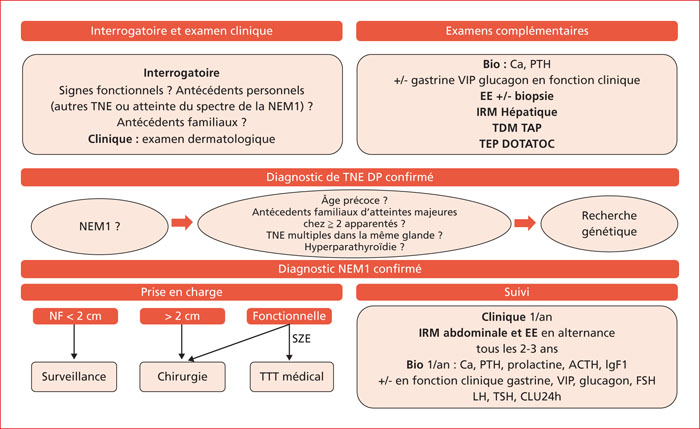
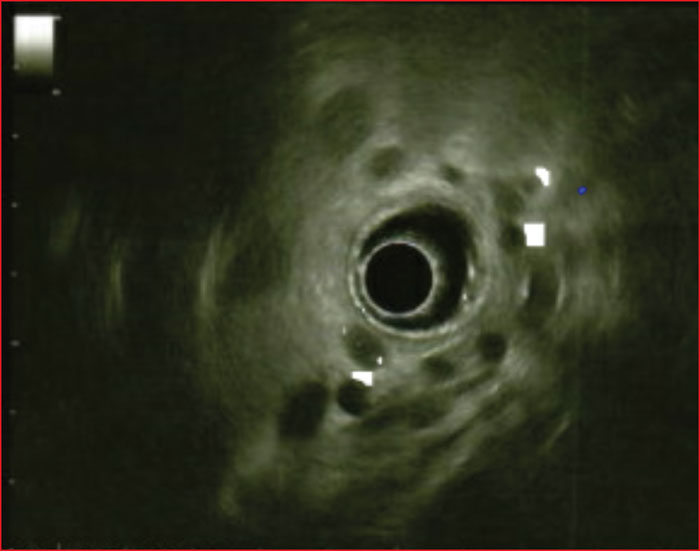
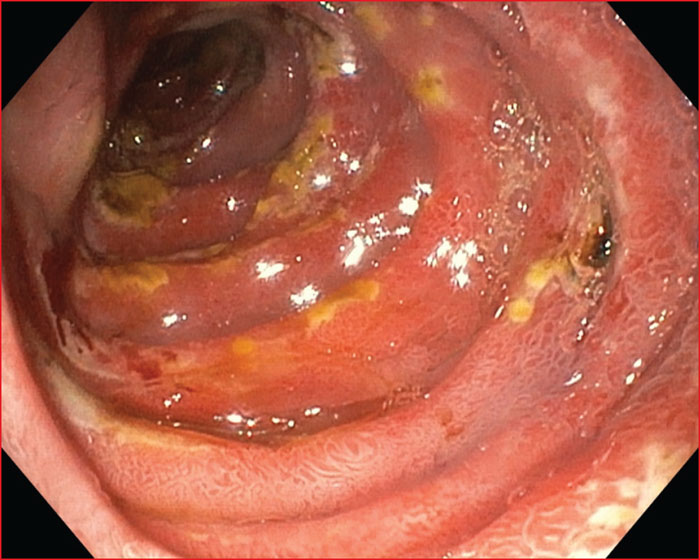
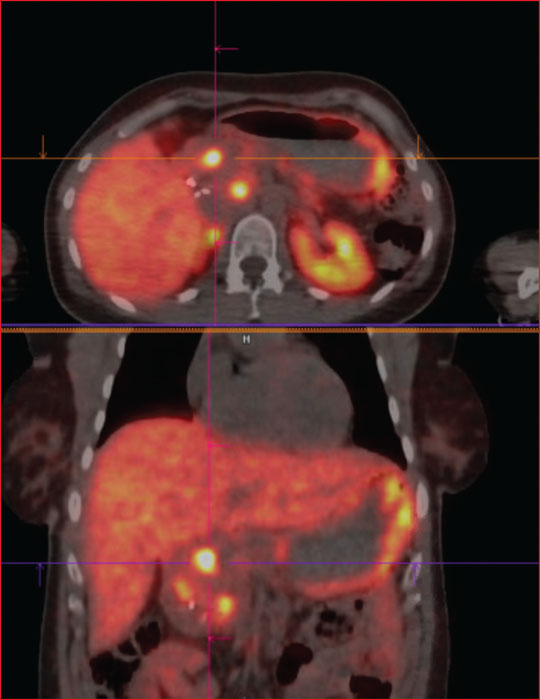
Multiple endocrine neoplasia type 1 (MEN1) is a rare, hereditary, autosomal dominant disease. It is characterized by the presence of cell hyperplasia or tumors of the endocrine system : parathyroid, duodenopancreatic, thymic, and bronchial neuroendocrine tumors (NETs), and pituitary and adrenal adenomas. Duodenopancreatic involvement is common and accounts for 60% of the causes of death in MEN1. Duodenopancreatic NETs can be functional or non-functional, benign or malignant. The most common functional duodenopancreatic NETs are gastrinomas and insulinomas. Patients with MEN1 require multidisciplinary management (gastroenterologist, endocrinologist, pathologist, radiologist, nuclear physician, surgeon) in an expert center and lifelong follow-up. Failure to diagnose MEN1 is a loss of opportunity for the patient and his family. This review is centered on duodenopancreatic NETs in MEN1, specifically diagnostic and therapeutic management.