Epileptic Disorders
MENUMyoclonic status epilepticus as a presentation of caspr2 antibody-associated autoimmune encephalitis Volume 16, issue 4, December 2014

Antibodies targeting the voltage-gated potassium channel (VGKC) complex and associated proteins have been identified as being relevant in both peripheral and central autoimmune neurological disorders, with autoimmune encephalitis now a recognised manifestation. We describe a patient with autoimmune encephalitis and antibodies targeting contactin-associated protein-like 2 (caspr2) who presented with myoclonic status epilepticus. To our knowledge, myoclonic status epilepticus has not been previously described with autoimmune encephalitis associated with caspr2 antibodies. Refractory status epilepticus and myoclonic status epilepticus are frequently associated with a poor outcome including death or severe neurological disability (Jumao-as and Brenner, 1990; Ferlisi and Shorvon, 2012). This case demonstrates that autoimmune encephalitis is an important differential diagnosis in such patients, and that early diagnosis and targeted immunotherapy can improve prognosis and functional outcome.
Case study
A 35-year-old woman presented with a three-day history of a viral prodrome with lethargy, myalgia, vomiting, and a non-specific headache. She became hypersomnolent, and developed recurrent prolonged generalised convulsions prompting admission. She did not have any risk factors for epilepsy. There was a strong family history of autoimmunity and she had a brother diagnosed with idiopathic thrombocytopenic purpura, a mother with Graves disease, and a maternal uncle with systemic lupus erythematosus.
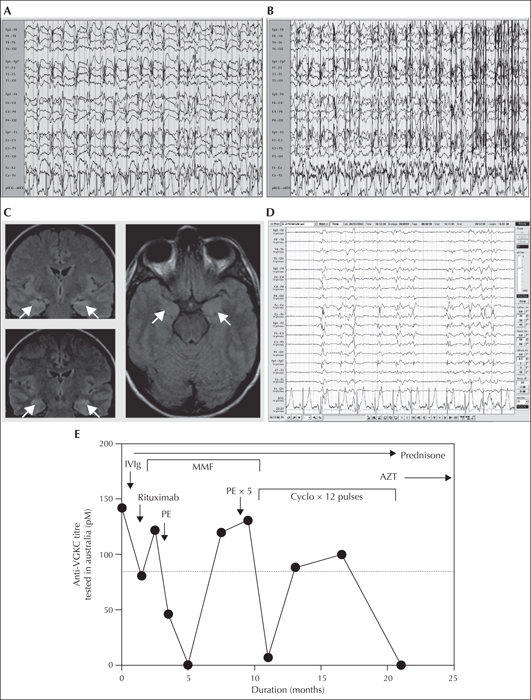
Initial observation revealed irregular bilateral myoclonus. She had prominent and asymmetric facial myoclonus, with eyelid, mouth, and tongue involvement, more notable on the left. She also had action myoclonus of the limbs. Progressive unresponsiveness prompted intubation for airway protection and a transfer to the intensive care unit. She continued to have refractory myoclonus (video sequence 1). Electroencephalograms (EEGs) revealed generalised periodic spike or polyspike-and-wave discharges at 2 Hz (figure 1A). Attempts at weaning sedation resulted in generalised myoclonic seizures without regaining consciousness (figure 1B).
Repeated cerebrospinal fluid (CSF) analyses demonstrated normal protein levels, a lymphocytic pleocytosis (44 × 106/l mononuclear cells), and intrathecal oligoclonal bands. CSF analysis was negative for herpes simplex virus (HSV), enterovirus, and Whipple's. A vasculitic screen, anti-phospholipid antibodies, and anti-thyroid antibody testing was negative. Serum and CSF antibodies against the N-methyl-D-aspartate receptor (NMDAR) and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR), and anti-neuronal antibodies against intracellular antigens, were negative. Magnetic resonance imaging of the brain demonstrated subtle bilateral hippocampal/parahippocampal high T2 fluid attenuated inversion recovery signal and mild diffusion restriction, without accompanying contrast enhancement (figure 1C). There was no malignancy on whole body positron emission tomography, and no thymoma on imaging. Needle electromyography did not reveal neuromyotonia.
A thiopentone coma was commenced to achieve burst suppression as treatment of status epilepticus, and coma was maintained with propofol and midazolam. Treatment with combinations of multiple anticonvulsants including phenytoin, levetiracetam, sodium valproate, topiramate, ethosuxamide, lacosamide, and clobazam failed to achieve seizure control without propofol and midazolam infusions. Repeated EEGs demonstrated diffuse cortical dysfunction, as well as bursts of maximal central discharges in synchrony with her clinical myoclonus (figure 1D). Frequently, clinical myoclonus was not accompanied by epileptiform discharges. Initial treatment included intravenous acyclovir and antibiotics on presentation until her HSV PCR and CSF cultures were proven to be negative. From the second day of admission, she received three doses of pulsed steroids (1 g/day) followed by oral prednisone (60 mg daily), and five doses of intravenous immunoglobulin to target a possible autoimmune encephalitis (figure 1E).
Serum antibodies against the VGKC complex were detected in pre-treatment admission blood tests. An initial sample tested at the Oxford laboratory demonstrated a VGKC antibody titre of 909 pmol/l (normal range 0-100). A cell-based assay demonstrated high titre antibodies targeting caspr2, with no antibodies present against leucine-rich glioma inactivated 1 (LgI1). Glutamic acid decarboxylase (GAD) antibodies were also elevated at 148 U/ml (normal range: 0-5). Due to refractory myoclonic status epilepticus, a ketamine infusion was trialed based on previously reported success in status epilepticus (Pruss and Holtkamp, 2008). Two doses of 1 g rituximab over a fortnight were also administered. Serial samples of VGKC antibody titres throughout the patient's disease course were also tested at a national laboratory (figure 1E).
Ten days after commencement of rituximab and with ongoing reduction in sedation, her level of consciousness improved and she was able to follow one-step commands. She had marked weakness secondary to deconditioning, and persisting action induced facial and limb myoclonus. Despite clinical stability, anti-VGKC complex titres continued to rise, prompting further treatment with five plasma exchanges and commencement of mycophenolate mofetil. She was transferred to an inpatient rehabilitation unit after a four-month admission, and discharged home one month later. Anti-VGKC antibody titres progressively declined and eventually become undetectable (figure 1E). On discharge, there were no further clinical or EEG seizures, but there was persisting facial, bulbar, and tongue myoclonus, as well as speech dyspraxia and dysarthria. Maintenance immunosuppression with mycophenolate 1 g bd and conservatively weaned oral prednisone was continued. Anticonvulsant therapy was rationalized to levetiracetam at 1 g mane/2 g nocte, carbamazepine at 600 mg tds, and clonazepam at 6 mg qid.
She experienced five months of clinical stability. However, persistent myoclonus, despite the absence of clinical seizures, and a rebound of VGKC levels, despite ongoing B cell depletion, prompted cessation of mycophenolate and institution of five further plasma exchanges and monthly pulses of cyclophosphamide over 12 months (500 mg/month). Twenty months post disease onset, she was seronegative for VGKC complex antibodies, with improved swallowing function, and no spontaneous myoclonus, although she had residual action-induced facial myoclonus and speech impairment (video sequence 2). Her functional improvement allowed a return to employment, with neuropsychiatric assessment demonstrating no significant residual cognitive deficits. Maintenance immunotherapy included 10 mg of prednisone and 75 mg of azathioprine daily.
Discussion
The VGKC complex is a diverse group of signalling proteins with multiple transmembrane subunits as well as auxiliary proteins that play a vital role in neuronal excitability (Vincent et al., 2004). Antibodies targeting the VGKC complex have been identified as being relevant in both peripheral and central autoimmune neurological disorders. Limbic encephalitis (LE) refers to the subacute onset of amnesia and behavioural changes often associated with sleep disturbance, seizures, and hallucinations, accompanied by medial temporal lobe signal change on MRI (Vincent et al., 2004). A number of patients with LE who have antibodies targeting cell surface receptors such as the VGKC complex, do not always have an associated malignancy, and are potentially treatment-responsive with immunotherapy, such as steroids, intravenous immunoglobulin, and plasma exchange (Vincent et al., 2004; Lancaster et al., 2011).
Unlike the other assays used to detect neuronal surface antibodies, the VGKC complex assay does not involve direct transfection of the antigen into a cell line. Instead, a complex of VGKC, which includes closely associated proteins, is bound using dendrotoxin. Antibody binding is detected using immunoprecipitation methods (Ramanathan et al., 2014). While it was initially thought that the antibodies targeted components of the VGKC itself such as various Kv1 subunits, it is now known that the majority of antibodies actually target proteins such as leucine-rich glioma inactivated 1 (LGI1) and contactin-associated protein 2 (caspr2), which are closely associated with the VGKC complex (Irani et al., 2010). For assays to detect caspr2 and LGI1 specifically, transfected cell lines are often used as a method to detect antibodies bound to these expressed proteins. While some overlap exists, LGI1 antibodies have been more frequently associated with patients presenting with LE and epilepsy without underlying malignancies, while antibodies targeting caspr2 have been associated with Morvan's syndrome, peripheral nerve hyperexcitability, and associated thymomas (Irani et al., 2010).
This case describes a presentation with myoclonic status epilepticus, which has not previously been associated with caspr2 antibodies. While the elevated level of GAD antibodies in this patient is noted, it is known whether this phenomenon is relatively common in autoimmune neurological disease and whether it may occur concurrently with other neuronal antibodies (Graus et al., 2010). Myoclonic status epilepticus in adults is classically associated with anoxic brain injury, and less commonly with metabolic encephalopathies and poorly controlled generalised epilepsy syndromes (Jumao-as and Brenner, 1990). Myoclonic status epilepticus, particularly in the case of anoxic brain injury, usually confers an extremely poor prognosis (Jumao-as and Brenner, 1990; Ferlisi and Shorvon, 2012). Our patient had a clinical presentation of new-onset encephalopathy and seizures, with CSF lymphocytic pleocytosis and unmatched intrathecal oligoclonal bands, supportive of a diagnosis of autoimmune encephalitis, and further substantiated by the detection of serum caspr2 autoantibodies. The improvement of her clinical course with immunosuppressive therapy mirrored by a reduction in the levels of serum antibodies, suggests, but does not confirm, a causative association. A case of opercular myoclonic-anarthric status epilepticus in association with GAD antibodies was recently reported (Monnerat et al., 2013), and highlights the notion that some cases of myoclonic status epilepticus may have an autoimmune aetiology amenable to immunotherapy.
This case demonstrates the essential role of immunosuppressive therapy in seizure control in patients with autoimmune encephalitis. Autoantibodies targeting cell surface receptors and transmembrane domains have been recognised as an aetiological cause of new-onset seizures in “autoimmune epilepsies” (Bien, 2013). Such patients have a reasonable chance of seizure freedom with the commencement of appropriate immunotherapy, but frequently fail to achieve seizure control on anticonvulsants alone (Bien, 2013). While there are no definitive therapeutic guidelines for the treatment of autoimmune encephalitis or epilepsy, recommended treatment often includes early therapy with intravenous immunoglobulin and/or pulsed intravenous methylprednisolone followed by oral prednisone, with failure to respond within a few weeks prompting consideration of plasmapheresis, or second-line therapy with broader immunosuppressive agents including cyclophosphamide or rituximab (Quek et al., 2012; Lancaster et al., 2011; Ramanathan et al., 2014).
This case highlights the fact that new-onset status epilepticus and myoclonic status epilepticus may not be universally poor predictors of outcome, and that autoimmune encephalitis needs to be considered as an early differential diagnosis in patients who present in this manner. Recognition is important as early diagnosis and targeted immunotherapy can improve seizure control, neurological recovery, and cognitive outcome.
Acknowledgements and disclosures
We acknowledge the assistance of Pathology Queensland (Queensland Health Clinical and Statewide Services) and Professor Angela Vincent's laboratory at Oxford, UK, for performing our assays. No funding was required for the work presented in this manuscript.
None of the authors have any conflicts of interest to disclose.