Epileptic Disorders
MENUKCTD7-related progressive myoclonus epilepsy Volume 18, supplement 2, September 2016

In 2007, three siblings from a consanguineous family with a clinical picture of progressive myoclonic epilepsy (PME) (Minassian et al., 2016) were reported, associated with a homozygous mutation of the gene encoding potassium channel tetramerization domain-containing protein 7 (KCTD7) (Van Bogaert et al., 2007). Since this original family, 16 other patients from 11 new families have been reported (Blumkin et al., 2012; Kousi et al., 2012; Krabichler et al., 2012; Staropoli et al., 2012; Farhan et al., 2014). The disease is inherited as an autosomal recessive trait and its incidence is unknown. In a study in which KCTD7 was screened in a cohort of more than 100 unconfirmed PME patients after exclusion of neuronal ceroid lipofuscinosis (NCL), five positive families were identified, suggesting that KCTD7 is not an exceptional cause of PME (Kousi et al., 2012). In a pilot study evaluating a panel of 265 genes, including KCTD7, in 33 index patients with various epileptic syndromes randomly selected in Germany and Switzerland, one patient was shown to have a homozygous mutation for KCTD7 (Lemke et al., 2012). The present article focuses on the clinical characteristics of KCTD7-related PME from the 19 published cases reported so far and discusses the pathophysiology and genotype/phenotype correlation of the disease.
Clinical characteristics
The 12 families reported so far (Van Bogaert et al., 2007; Blumkin et al., 2012; Kousi et al., 2012; Krabichler et al., 2012; Staropoli et al., 2012; Farhan et al., 2014) had variable ethnic origin and showed a high rate of consanguinity (5/12). Epileptic seizures were the first signs in all cases. Onset ranged between five months and three years, after a period of psychomotor development that was described as either normal or slightly delayed. Independent walking was usually acquired. However, one reported patient with onset in the second year of life never walked (Kousi et al., 2012). The first seizures were described as either myoclonic or generalized tonic-clonic seizures, and were precipitated by fever in some cases. Myoclonic seizures were reported in the course of the disease in all but one patient. In 11 patients, continuous multifocal myoclonus, aggravated by action and posture, was reported either at presentation or during the course of the disease. In one patient, myoclonus was associated with opsoclonus, which prompted the authors to consider a diagnosis of opsoclonus-myoclonus syndrome (Blumkin et al., 2012). Other types of seizures (atonic seizures and atypical absences) were also reported. Epilepsy responded poorly to antiepileptic drugs in 2/3 of patients, the most effective drugs being levetiracetam, valproate and clobazam. All patients had cognitive decline leading to severe dementia. Progressive ataxia was reported in all but one patient, and two thirds of them became wheelchair-bound. Pyramidal signs were described in eight patients and microcephaly in six patients.
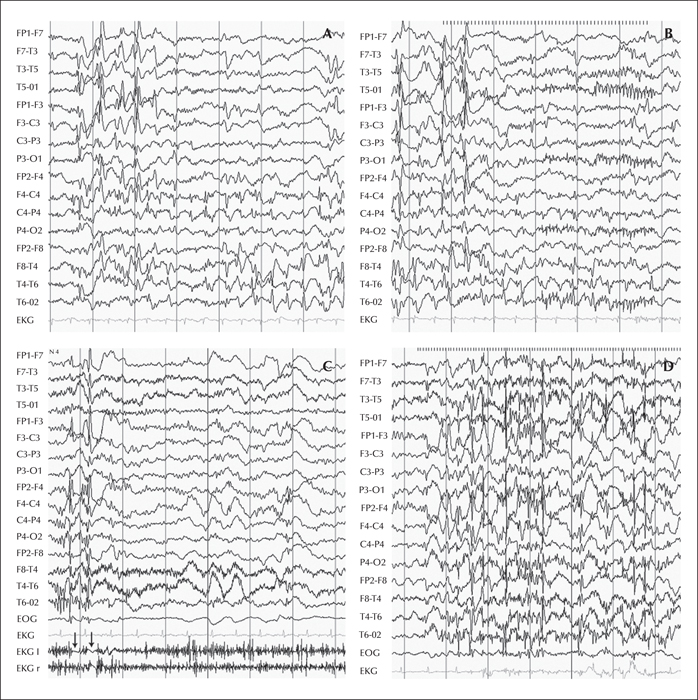
Concerning complementary examinations, EEGs were described to be abnormal, except for those very early during the course of the disease. The most common findings were very frequent multifocal and/or generalized spike-waves associated with an excess of slow activity. In a number of patients, EEG abnormalities were more prominent in the posterior areas, and intermittent light stimulation evoked generalized or posterior epileptiform discharges. In some patients, the EEG was reported as either hypsarrhythmic or showing continuous spike waves during slow-wave sleep. Action myoclonus was not associated with concomitant epileptiform discharges. One patient had negative focal myoclonus. Figure 1 illustrates EEG findings from two affected siblings from the first family (Van Bogaert et al., 2007).
Cerebral magnetic resonance imaging was either normal or showed non-specific findings (atrophy or posterior white matter hyperintensities). Metabolic work-up of blood and cerebrospinal fluid was not suggestive of mitochondrial dysfunction. Fundoscopy was normal in most patients.
The search for storage material was performed by skin biopsy in nine patients from eight different families and was normal except for one patient who was reported to have electron-dense storage material in fibroblasts and a lymphoblastoid cell line based on electron microscopy (Staropoli et al., 2012). This finding is noteworthy as it was the only finding of storage material in a patient mutated for KCTD7, which prompted the authors to designate KCTD7 mutations as the cause of a new subtype of NCL that was called ‘NCL type 14’. It should be noted that the screening for KCTD7 mutation performed in a cohort of 22 NCL patients with storage material demonstrated by electron microscopy analysis was negative, suggesting that KCTD7 is not a common cause of NCL after exclusion of the already known NCL genes (Kousi et al., 2012). Moreover, the patient reported by Starapoli et al. (2012), as well as his affected brother, had a very atypical clinical course that differed from other KCTD7 patients. First, these 2 patients were the only reported cases with severe ocular involvement, i.e. optic atrophy leading to visual loss. Second, constant clinical deterioration was observed in these 2 patients who died in their mid-teens. This contrasted with the other reported KCTD7 patients who showed clinical stabilization after a period of early regression, most of whom were still alive at the last assessment.
To our knowledge, the oldest patient (Patient 2 from the original report [Van Bogaert et al., 2007]) is now aged 25 years and is still able to walk independently.
Differential diagnosis
The typical presentation follows a 3-stage evolution that is quite unique among PME:
- (1)a period of normal development;
- (2)a period of mental and motor regression with epileptic myoclonic seizures that starts at around 1-2 years of age;
- (3)and a period of stabilization after a few years.
Rare cases with optic atrophy and storage material on skin biopsy may complicate differential diagnosis with the classic late-infantile form of NCL. Clues to resolve this differential diagnosis are summarized in table 1. The rare presence of associated opsoclonus can evoke opsoclonus-myoclonus syndrome. Finally, in rare patients, ataxia or myoclonus may not be present, which raises the issue of whether such patients have an epileptic syndrome other than PME.
Physiopathology
All 11 different mutations identified so far are within the coding region of KCTD7, which is a member of the KCTD gene family. This family of proteins shares an N-terminal BTB/POZ domain that demonstrates sequence homology to the T1 domain in voltage-gated potassium channels that allows its tetramerization (Stogios et al., 2005). The KCTD7 protein is extremely conserved across species and, in mice, is expressed in the olfactory bulbs, the CA1 and CA3 hippocampal cells, and the Purkinje cells of the cerebellum (Azizieh et al., 2011). KCTD7 overexpression in transfected primary cultures of murine neurons resulted in hyperpolarization of the resting membrane potential, and decreased their excitability in patch clamp experiments (Azizieh et al., 2011). This is consistent with an epileptogenic effect of a KCTD7 defect. KCTD7 is much smaller than a typical potassium channel subunit, and its computed hydrophobicity profile does not indicate a transmembrane segment. It is hence extremely unlikely that KCTD7 would function as a transmembrane channel for potassium. The demonstration of a direct molecular interaction between KCTD7 and Cullin 3 suggests that the effect on the plasma membrane resting potential is likely to be mediated by Cullin 3 (Azizieh et al., 2011). Although additional experiments are required to understand how the KCTD7 defect causes PME, this may already explain the lack of genotype/phenotype correlation observed in affected patients. Indeed, mutations within the functional BTB/POZ domain (5/11 reported mutations) did not result in a more severe phenotype.
An interesting finding is that the severity of the disease may be highly variable among affected members within the same family (Van Bogaert et al., 2007; Farhan et al., 2014). Since the more severely affected members also had a more severe epileptic disorder, this suggests that the epileptic activity itself played a role in the neurological deterioration observed in patients. From this point of view, KCTD7-related epilepsy could be considered as an epileptic encephalopathy, i.e. a condition in which epileptic activity may contribute to progressive neurological decline (Berg et al., 2010), such that effective antiepileptic intervention might improve developmental outcome.
Disclosures
The author has no conflict of interest to disclose.
The present manuscript is part of a Epileptic Disorders/Mariani Foundation supplement on Progressive Myoclonus Epilepsies, downloadable as a whole by visiting www.epilepticdisorders.com.