Epileptic Disorders
MENUEpilepsy after cerebral infection: review of the literature and the potential for surgery Volume 19, issue 2, June 2017

Cerebral infection is a frequent cause of symptomatic focal epilepsy, particularly in the developing world (Singhi, 2011; Ngugi et al., 2013) and the most common preventable cause of epilepsy worldwide (Sander and Perucca, 2003). Several risk factors for the development of chronic epilepsy after cerebral infection have been identified. Despite the high rates of pharmacoresistance in epilepsy after cerebral infection, especially in patients with structural brain lesions as sequelae of the infection, the referral is low, and this aetiology is under-represented in epilepsy surgery series. However, surgical outcomes in certain subgroups of patients with epilepsy after cerebral infection are encouraging, whereas constellations with less favourable seizure outcomes are readily identifiable.
This review derives from a comprehensive literature search using PubMed to identify the prevalence and characteristics of epilepsy after cerebral infection, as well as the significance of epilepsy surgery in this context. The literature search included English-language full-length articles published between September 1988 and July 2016. We also searched bibliographies of original articles, review articles, and book chapters to identify relevant studies. We used combinations of the following key words: infection, encephalitis, meningitis, post-infectious, post-encephalitic, post-meningitic, epilepsy, epilepsy surgery, risk factors, outcome, prognosis, herpes simplex, and neurocysticercosis (NCC).
The review is divided into two parts: Part I deals with epilepsies after cerebral infection in a more general overview. Part II deals with the particular features of epilepsies caused by different infectious agents.
Part I: overview of epilepsies after cerebral infection
Development of epilepsy after cerebral infection
Late unprovoked seizures occurring months to years after the insult reflect epileptogenesis (Pillai et al., 2016) and should be distinguished from early (acute) symptomatic seizures, occurring within the first two weeks after a cerebral infection in one third of cases (Singhi, 2011). Overall, the reported risk of late unprovoked seizures in population-based cohorts of cerebral infection survivors from developed countries is estimated at 7-8% and is much higher in resource-poor countries (Vezzani et al., 2016). In a population-based study from the USA, the risk of developing unprovoked seizures after acute encephalitis or meningitis was 2% in the first year, rising to 7% twenty years after the acute infection. The highest incidence of unprovoked seizures occurred in the first five years (Annegers et al., 1988).
Not only is the incidence and prevalence of epilepsy after cerebral infection considerably higher in the developing world, but the spectrum of associated infectious agents is also much larger compared to developed countries (Singhi, 2011). “Neuronal infection” was identified as the aetiological substrate in 22% of 719 consecutive children with epilepsy who attended a tertiary paediatric centre in Mexico City over a period of four months (Ruiz-García et al., 2002). Of 346 patients with symptomatic epilepsies seen at a University Hospital in Western China, 12% of patients aged <18 years and 25% of patients aged 18 to 60 years had epilepsy after cerebral infection. Aetiology included viral encephalitis (VE) in 38% of cases, NCC in 34%, and tuberculosis in 25% (Si et al., 2012). Cysticercosis cellulosae is one of many parasites, such as plasmodium falciparum, toxoplasma gondii, Schistosoma japonicas, and toxocara caniis, causing seizures and subsequently epilepsy. Epilepsy after cerebral infection with these other parasites is, however, rarer compared to NCC.
The main risk factors for epilepsy after cerebral infection are:
- –status epilepticus during the acute phase;
- –the infectious agent;
- –and parenchymal brain lesions as a result of the infection (Lee et al., 2007).
Further features predictive of epilepsy after cerebral infection included the age at acute infection, the severity of brain injury, and the genetic substrate (Michael and Solomon, 2012). Status epilepticus, a life-threatening neurological and medical emergency with high mortality and morbidity (Raspall-Chaure et al., 2006), has been shown to constitute a major risk factor for drug-resistant epilepsy after cerebral infection (Fowler et al., 2010; Pillai et al., 2016). Status epilepticus is more frequent in countries with limited healthcare resources, thus contributing to the higher incidence and prevalence of epilepsy after cerebral infection in developing countries. Sadly, the frequency of status epilepticus following cerebral infections in these countries can be three times higher in children compared to adults (Singhi, 2011).
The mechanisms by which different infectious agents produce acute seizures and then later on unprovoked seizures have been recently addressed in detail (Vezzani et al., 2016).
Prevalence and predictors of drug-resistance
In a multicentric French study (19 paediatric and four adult tertiary epilepsy centres) including 5,794 patients with epilepsy reported to the French National Epilepsy Register Group (GRENAT), 128/5794 (3%) had epilepsy after cerebral infection (Chipaux et al., 2016). Of these, 90/128 (70%) were characterized as focal, corresponding to 7% of 1,280 focal epilepsies in this database; 40% were drug-resistant. In a multicentric Italian study (11 tertiary centres) including 1,124 patients (933 adults, 191 children) with refractory epilepsy, 11/191 (6%) children and 73/933 (8%) adults had epilepsy caused by “postnatal infections and other postnatal factors” (Alexandre et al., 2010). Epilepsy after cerebral infection was the second most frequent aetiology (15%) in 199 children with focal epilepsy admitted to the Japanese national epilepsy centre in Shizuoka in the years 1993 and 1994 (Fujiwara and Shigematsu, 2004). This particularly high prevalence of epilepsy after cerebral infection compared to both multi-centric studies may be attributed to a sample bias, considering that the report from Shizuoka refers exclusively to children and that children with severe epilepsy may be over-represented in this national epilepsy centre. Almost half (14/30) of these children presented pharmacoresistance.
In a recently reported acute encephalitis cohort with 147 patients followed for 2-15.8 years (Pillai et al., 2016), 21% developed epilepsy after cerebral infection; 10% were drug-resistant. Status epilepticus was the only independent predictor of pharmacoresistance. Further predictors of pharmacoresistance in epilepsy after cerebral infection include:
- –focal seizures;
- –intensive care;
- –three or more AEDs;
- –epileptic discharges on EEG;
- –T2/ FLAIR hyperintensity of the mesial temporal structures and gadolinium enhancement on MRI (Lee et al., 2007; Fowler et al., 2010; Rismanchi et al., 2015 ; Pillai et al., 2016).
Interestingly, poor response to AEDs correlated with herpes simplex virus (HSV) infections (33%), but not with acute disseminated encephalomyelitis (ADEM), enterovirus, or anti-N-methyl-D-aspartate receptor-(NMDAR) encephalitis (Pillai et al., 2016). Moreover, the gadolinium enhancement is a typical MRI feature of herpes simplex virus encephalitis, previously linked to a severe postencephalitic course (Fowler et al., 2010). These observations underline the multifactorial determination of pharmacoresistance in epilepsy after cerebral infection.
It should be noted that the vast majority of pharmacoresistant epilepsy after cerebral infection relates to an MRI/CT-identifiable structural lesion (Wirrell et al., 2011; Chipaux et al., 2016), which also applies to non-parasitic aetiologies. In the Connecticut community-based study, 613 children aged one month to 16 years were enrolled between 1993 and 1997 (Berg et al., 2009). In total, 518/613 (85%) had “usable MRI scans”. Structural lesions were found in 82/518 (16%). Only three lesions due to infectious disease were reported: one was bi-hemispheric, one was located in the thalamus, and another was located in the temporal lobe and attributed to postnatal toxoplasmosis. This last case was the only case of epilepsy after cerebral infection considered for epilepsy surgery.
Surgery for epilepsy caused by cerebral infection
Patients undergoing surgery for drug-resistant epilepsy after cerebral infection constitute only a small minority in epilepsy surgery series. In the 2004 international survey of paediatric epilepsy surgery (Harvey et al., 2008), the neuropathological diagnosis of “chronic encephalitis”, including Rasmussen encephalitis, concerned only 10 (2%) of 458 cases. Of 9,576 cases registered in the European Epilepsy Brain Bank (EEBB) (Blümcke et al., 2016), only 80 (0.8%) are listed under “encephalitis”, excluding Rasmussen encephalitis patients. However, epilepsies linked to mesial temporal sclerosis following encephalitis or meningitis are listed in the EEBB under this neuropathological diagnosis alone.
Precise rates of epilepsy after cerebral infection in each epilepsy surgery centre are hard to obtain. In particular, major epilepsy surgery centres hardly ever publish their experience concerning all aetiologies, and if they do so, they tend to lump together reports about surgeries for epilepsy after cerebral infection with other, rarer aetiologies (Hemb et al., 2010). Of 120 children operated on between 1998 and 2009 at the Claudio Munari epilepsy surgery centre in Milano, Italy, nine had a pathology labelled as “flogistic” (Teutonico et al., 2013). No patients with epilepsy after cerebral infection were listed in a report on 107 children operated on at the epilepsy centre Ribeirao Preto, Brazil, between 1994 and 2002 (Terra-Bustamante et al., 2005); two were classified as “miscellaneous”. In the Canadian survey on epilepsy surgery in the first 3 years of life (Steinbok et al., 2009), 6/116 (5%) patients had an “encephalitis” (“including one TORCH-infection”): two had an Engel I outcome, two Engel II, one each Engel III and IV.
In a comprehensive review of presurgical evaluation and surgical outcome in drug-resistant epilepsy after cerebral infection, only five relevant papers published between 1970 and 2011 were identified (Sellner and Trinka, 2013). Two studies focused on preoperative evaluation and on postsurgical seizure outcome in epilepsy after BM (Davies et al., 1996; Trinka et al., 2000). Three studies include both epilepsy after BM and after VE (Lee et al., 1997; O’Brien et al., 2002; Donaire et al., 2007) (table 1).
Temporal resections
The vast majority of patients who have undergone temporal resections feature drug-resistant epilepsy linked to mesial temporal sclerosis and are thus candidates for anterior temporal lobectomy, with excellent results (Sellner and Trinka, 2013). The association between epilepsy after cerebral infection and mesial temporal sclerosis had already been identified in the early 90s (Marks et al., 1992; Lancman and Morris, 1996). In a study including both meningitis (n=16) and encephalitis (n=22) patients, post-meningitic epilepsy was commonly associated with unilateral mesial temporal sclerosis, whereas most encephalitis patients featured additional neocortical foci. Interestingly, the age at cerebral infection was crucial for the emergence of mesial temporal sclerosis and thus for the development of mesial temporal epilepsy: only cases of encephalitis before age 4 were associated with mesial temporal sclerosis. This pointed to a window of vulnerability for the hippocampus. Thus, the spectrum of post-encephalitic epilepsy contains unilateral mesial temporal sclerosis cases, as well as multifocal (neocortical) epilepsy due to widespread brain injury (Davies et al., 1996).
Extratemporal resections
Extratemporal resections are rarely performed in patients with drug resistant epilepsies due to cerebral infections, notwithstanding the few reports on surgery for drug-resistant epilepsy due to parasitic brain infections. In a meta-analysis of paediatric extratemporal epilepsy surgery, pooling together 1,259 children from 36 studies published in 1993-2012 (Englot et al., 2013), only seven children had an “infection” as the underlying aetiology. These patients had a less favourable outcome compared to most other aetiologies: 35% Engel I, 65% Engel II-IV. The lower rates of extratemporal surgery for epilepsy after cerebral infection may reflect the limitations of surgical treatment in the context of multifocal epilepsy, particularly for post-encephalitic cases arising from brain damage beyond the mesial temporal structures.
Hemispheric resections/disconnections
Patients with drug-resistant hemispheric epilepsy and contralateral hemiparesis as a result of a severe hemispheric injury are candidates for hemispherectomy/hemispherotomy. From a conceptual viewpoint, patients with epileptogenic hemispheric damage caused by status epilepticus during the acute cerebral infection may be perceived as suffering from hemiconvulsion-hemiplegia-epilepsy (HHE) syndrome. “Early inflammation” was given as the aetiology in 5/17 patients (four with epilepsy after VE and one with epilepsy after BM) who underwent hemispherectomy in Minneapolis, USA in 1950-1971, and were followed for over 38 years (Davies et al., 1993). Three of these patients remained seizure-free, one had a single seizure 16 years after surgery, and one had a 50% seizure reduction. Although the acute cerebral infection and subsequent seizure onset occurred in the first year of life in 4/5 patients, hemispherectomy was performed with considerable delay at age 10, 11, 13 and 19 years, with excellent results in three cases. “Post-encephalitis/post-meningitis” was the cause of drug-resistant epilepsy in 17/115 (15%) patients who underwent hemispherectomy/functional hemispherectomy in the UCLA, USA from 1986 to 2002 (Jonas et al., 2004): no information is provided regarding seizure outcome in this particular subgroup. In a study from the Great Ormond Street Hospital in London, Great Britain, 2/33 patients presenting “postnatal encephalitis” underwent hemispherectomy between 1991 and 1997 (Devlin et al., 2003): again, no information is given regarding seizure outcome in this particular subgroup. In the largest monocentric (Cleveland Clinic, USA) hemispherectomy series to date, including 170 patients (Moosa et al., 2013a, 2013b), no distinct aetiological group, such as “post-inflammation”, “post-infectious disease”, or “post-encephalitis/post-meningitis”, is identified. Patients with epilepsy after cerebral infection may be listed under the aetiological group of “encephalomalacia” (79/170), but are apparently very few. The group of “encephalomalacia” includes “various aetiologies, such as remote ischaemic stroke, prior intracerebral haemorrhage, asymmetric hypoxic-ischaemic injury, and prior head trauma”, but does not explicitly contain epilepsy after cerebral infection. Finally, surgery for epilepsy caused by cerebral infection was lumped together with other rarer aetiologies in the category “others” in the large multicentric survey regarding “post-hemispherectomy seizures” (Holthausen et al., 1997). This category comprised only 28 (9%) out of 328 hemispherectomy/hemispherotomy patients.
Part II: Infectious agents and Epilepsy
Epilepsy after viral encephalitis
The main risk factors for epilepsy after viral encephalitis (VE) in 198 acute encephalitis patients in a study from the Mayo Clinic, USA (Singh et al., 2015) were:
- –seizures (particularly focal) during the acute phase of the disease;
- –status epilepticus;
- –and FLAIR/T2 hyperintensity on MRI, with “structural injury often involving the epileptogenic temporal and frontal lobes”.
Indeed, in an earlier population-based study (Annegers et al., 1988), the risk for the development of chronic epilepsy at 20 years of follow-up was 22% for patients with acute seizures compared to 10% for patients without acute seizures. Cortical injury, as for HSV-1 and less frequently HSV-2 infections, is apparently linked to higher epilepsy rates (Singh et al., 2016), compared to viral infections with damage mainly to subcortical structures, as in Japanese encephalitis (Lee et al., 2007; Misra et al., 2008). None of the patients with “pure subcortical involvement, leptomeningeal enhancement, and brain stem or spinal cord lesions” on MRI developed epilepsy after VE. Additional risk factors for chronic epilepsy after VE, as identified in a study from the Taiwan University Hospital in 54/330 (16%) children with acute encephalitis between 1984 and 2000 (Lee et al., 2007), were focal neurological signs, severe disturbance of consciousness, and neurological deterioration during the hospitalization. Epilepsy after VE presented within six months after the acute illness in 80% and within the following three years in 94% patients; 26 (50%) cases were drug-resistant.
Epilepsy after herpes simplex virus encephalitis
HSVencephalitis (HSVE) is the most common sporadic encephalitis (De Tiège et al., 2003). The estimated incidence is 1/250,000-500,000 per year with one third of cases occurring in childhood and adolescence; over 90% are caused by HSV-1 (Baringer, 2008). Epileptologists are more often confronted with epilepsies after HSVE than with epilepsies after encephalitis caused by other viral agents. For this reason, peculiarities of this virus that may be relevant to the development of chronic epilepsies are discussed in more detail.
In contrast to the lower mortality since the invention of acyclovir, morbidity is still high, with many affected persons subsequently suffering neurological deficits. Older age at the acute stage of disease, development of coma, the presence of restricted diffusion on brain MRI, and delay in the administration of acyclovir predicted poor outcome in 29/45 adult patients with HSVE admitted to the Mayo Clinic between 1995 and 2013 (Singh et al., 2016). In this study, 10/22 (45%) “for which this information was available” developed epilepsy during the 6-12-month follow-up. The risk of severe neurological sequelae is particularly high in very young children. In a community-based three-year prospective survey of children aged 2-23 months in Britain and Ireland (Ward et al., 2012), 13/19 children with HSV-encephalitis aged 2-11 months during the acute illness had focal abnormalities on MRI and 11/19 had long-term neurological deficits. Of the six children aged 12-35 months during the acute illness, only one had an abnormality on MRI; 3/6 suffered from neurological sequelae. After a follow-up period of over one year, 12/19 were diagnosed with developmental delay and 7/19 (37%) had epileptic seizures.
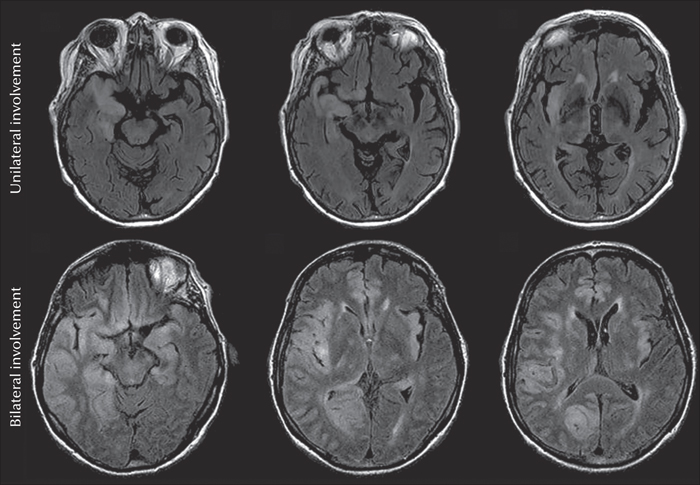
Due to the necrotizing properties of the HS viruses, epilepsy following HSVE is bound to arise from areas of MRI-visible cortical damage. Extratemporal regions may often undergo damage that is unfortunately also bilateral in many cases. At the early stages of HSVE, lesions arise from damage to the blood/brain barrier with consequent cerebral oedema, characterized by hypointensity on T1-weighted and hyperintensity on T2-weighted MRI. At later stages, an evolution towards haemorrhagic and necrotic lesions is evident. The localization and extent of T2/FLAIR signal changes reflecting the damage occurring in the acute stage of the disease were recently described in detail (Singh et al., 2016) (figure 1). In this cohort, T2/FLAIR signal changes were present in >90% of cases, with a slightly lower rate of cortical involvement. Changes were seen in the temporal lobes in 88%, in the insular cortex in 70%, in the frontal lobes in 68%, and in the thalamus in 28% of cases, whereas bilateral changes were diagnosed in almost half of the patients. These high rates of brain lesions are noteworthy since 52% of patients underwent acyclovir treatment from Day 1. The oedemas will eventually resolve in a significant number of patients. However, the distribution of acute MRI changes (figure 1) matches the distribution of cortical scars, subcortical white matter changes, and atrophic changes in patients with drug-resistant epilepsy following HSVE (Misra et al., 2008; Pillai et al., 2016; Singh et al., 2016). This observation underlines that temporal lobe epilepsy following HSV-encephalitis is rarely a “pure” temporal lobe epilepsy, and far more often a “temporal plus” epilepsy (Kahane et al., 2015; Barba et al., 2016).
Anti-NMDA-R encephalitis after HSV infection is a recently identified constellation that should be distinguished both from HSV reactivation and drug-resistant epilepsy after VE. NMDA-R antibodies have been frequently detected in patients with HSV-1 encephalitis (Prüss et al., 2012). Moreover, anti-NMDA-R encephalitis has been highlighted as a parainfectious autoimmune phenomenon, emerging within weeks of HSV-1 encephalitis (Armangue et al., 2013, 2014, 2015; Hacohen et al., 2014). Recent studies have substantiated previous suggestions that early relapse in HSV encephalitis, particularly in children, may be of autoimmune origin (De Tiège et al., 2003). The presentation is typically biphasic, with remission from VE, followed by relapse with autoimmune encephalitis within 1-7 weeks of first VE symptoms. Prompt diagnosis is crucial to initiate potentially beneficial immunotherapy (Armangue et al., 2015).
Epilepsy after cerebral viral infections other than HSV-encephalitis
The number of viruses that can cause encephalitis and subsequent chronic epilepsy is huge. Their worldwide geographical distribution is reported by Misra et al. (2008) (table 1) and discussed by Singhi (2011). In the Mayo Clinic report (Singh et al., 2014) on acute encephalitis pathogens (n=95), the following viruses were identified: HSV in 39%, varicella zoster in 23%, West Nile virus in 19%, Epstein-Barr virus in 6%, HIV in 3%, and other viruses in 10%. Factors associated with the development of epilepsy after VE were focal seizures, FLAIR/T2 abnormalities pointing to a cortical involvement, generalized seizures, and status epilepticus. Regarding epilepsy after VE, no statistically significant difference was found between HSV-encephalitis and encephalitis caused by other viruses in this study, although this has been shown in other publications (Singh et al., 2015). A similar spectrum of viruses, in cases with a positive result, has been found in the series from the Johns Hopkins Hospital in Baltimore, including 103 cases of acute encephalitis in adults (Thakur et al., 2013), although the cause was “unknown” in almost half of the patients. The aetiology remained unclear in 72% of encephalitis cases in a collaborative study with the participation of 17 hospitals in Spain (de Ory et al., 2013), as well as in 57% children with encephalitis in Taiwan (Lee et al., 2007). The significance of negative investigations for infectious agents at the time of the acute encephalitis in patients later developing drug-resistant epilepsies is discussed below under non-MTLE after viral encephalitis. The spectrum of viruses in the Taiwan study seems to be larger than in the studies from the US and Spain: enterovirus-encephalitis outnumbers varicella zoster and HSV infections, followed by infections caused by rubella, mumps, measles, and adenoviruses. In contrast to HSV, evidence for one of these other viruses was not associated with an increased risk of epilepsy after VE, notwithstanding the other known risk factors.
Epilepsy after VE is uncommon in Japanese encephalitis, the most frequent cause of encephalitis in Asia and Northern Australia. Though acute seizures tend to occur in up to 50% of affected persons (Misra et al., 2008), only 4% of patients from the Gansu province in China developed chronic epilepsy during (a relatively short) follow-up (Yin et al., 2015). Other neurological sequelae, such as various kinds of motor disturbances, dystonia and Parkinsonism, and cranial nerve symptoms, prevail along with impaired cognition and memory problems. This low rate of epilepsy after VE is due to the fact that Japanese encephalitis mainly affects the basal ganglia, the brain stem, and the spinal cord (anterior horn). Moreover, Japanese encephalitis often causes status epilepticus, which may lead to widespread cortical damage, with the subsequent development of drug-resistant epilepsy (seefigure 2ofMisra et al., 2008).
Surgery for epilepsy caused by viral encephalitis
With respect to surgical strategies for drug-resistant epilepsy after VE, different scenarios may apply, depending on the aetiology.
Surgery for epilepsy caused by HSV encephalitis
Lesions giving rise to epilepsy following HSVE often localize in brain regions unfavourable for epilepsy surgery. They are preferentially located in the perisylvian region, including the insula, and in the temporal lobes, though rarely confined to a circumscribed area in just one temporal lobe.
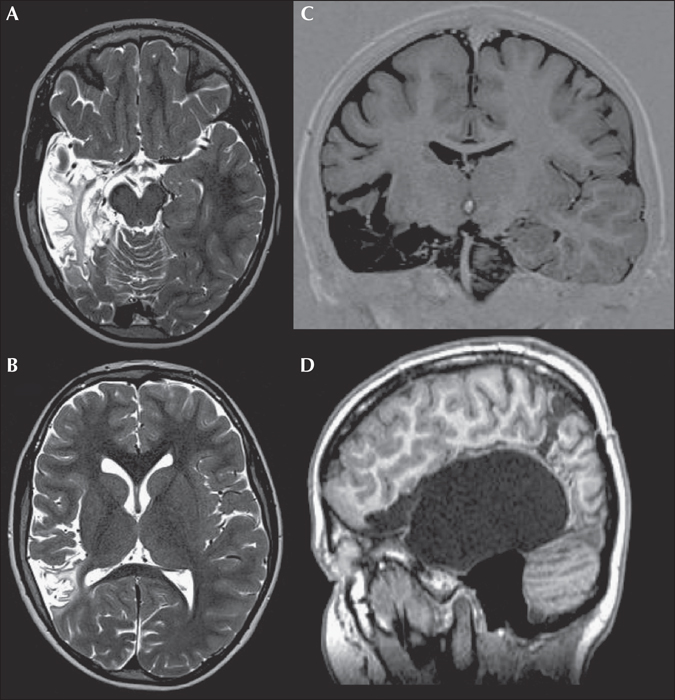
Only 1/4 patients with epilepsy following HSVE in a surgical series of patients with epilepsy after VE from the Montreal Neurological Institute became seizure-free (Trinka et al., 2000). Of children and adolescents operated on between 1999 and 2014 at the epilepsy centre in Vogtareuth, Germany, 5/430 (1%) underwent surgery for drug-resistant epilepsy following HSV-encephalitis. Of these, 3/5 patients had bilateral MRI and 2/5 had bilateral EEG abnormalities; 2/5 became completely seizure-free (Engel 1a; figure 2) and 3/5 had worthwhile improvement (Engel IIIa) (Weber et al., 2014). In fact, surgery in children with epilepsy after HSVE is often palliative, since the majority suffers from particularly severe bi-hemispheric extratemporal epilepsy. In this context, a palliative approach implies the resection of the leading focus or of the focus generating the most disabling seizure type. Moreover, emergency temporal lobe surgery may be performed in epilepsy after HSVE in order to stop a status epilepticus, a transtentorial herniation, or both (Sanchez-Carpintero et al., 2008; Safain et al., 2015; Bick et al., 2016).
Another issue related to cerebral HSV infections consists of HSV reactivation, a rare potential complication of epilepsy surgery in children with grave consequences. In all cases reported to date, symptoms appeared within the first two weeks following surgery. The most common presenting sign was fever, followed by focal or generalized seizures. In most cases, the MRI was diagnostic and revealed oedema, diffusion restriction, and/or abnormal enhancement in one or both temporal and frontal lobes, the insular cortex, and the angular gyrus (Bourgeois et al., 1999; Gong et al., 2010; de Almeida et al., 2015; Lo Presti et al., 2015).
Surgery for epilepsy caused by other viral agents - or with no identified virus at the time of acute encephalitis
No surgical series have been published regarding epilepsies caused by viruses other than HSV. In general, patients with epilepsy after VE caused by other viral agents or when no infectious agent was found can be subdivided into two groups:
- –patients with a history of encephalitis in early childhood who present, after a longer latency period, with mesial temporal lobe epilepsy (MTLE); and
- –patients with a history of encephalitis who present with epilepsies other than MTLE.
Patients presenting (1) a history of“encephalitis” in early childhood, (2) a longer “silent period” between acute infection/acute seizures and the onset of epilepsy, and(3)MRI evidence ofhippocampal atrophy, are excellent candidates for epilepsy surgery (Trinka et al., 2000; Donaire et al., 2007; Sellner and Trinka, 2012, 2013) (table 1). Seizure semiology and interictal and ictal EEG are compatible with the diagnosis of MTLE. The majority of these patients present cognitive functioning within the average range. In fact, a careful re-evaluation of their history often reveals a febrile episode in early childhood, during which one or a few seizures occurred, leading to the diagnosis of encephalitis based on clinical grounds alone, despite a negative screening for viral agents. In retrospect, parents almost always deny a condition of altered consciousness or lethargy in the acute phase of the disease, with or without personality changes, for over 24 hours, which constitute the main diagnostic criteria for encephalitis (Venkatesan et al., 2013). In these cases, hippocampal atrophy is most likely linked to the febrile convulsions.
In the epilepsy surgery study from Yale, USA, only patients with “encephalitis” before age 4 had unilateral MTLE and a favourable surgical outcome (Marks et al., 1992). In a report from Korea (Lee et al., 1997), 3/8 surgical cases with epilepsy after VE were younger than 7 years; at the latest follow-up visit, two had an Engel I and one an Engel II outcome. Of 36 patients who received surgery, with “catastrophic” epilepsy after VE in a study from the Montreal Neurological Institute, somewhat older during the acute infection (between age seven and ten), four patients had unilateral mesial temporal sclerosis, as well as atrophy in two cases; three had an Engel I and one an Engel II outcome (Trinka et al., 2000). It is important to keep in mind that young age at surgery is a predictor for unilateral MTLE and for favourable postsurgical seizure freedom only when associated with a longer “silent period” between the acute infection and the onset of epilepsy. In other words, in patients with hippocampal sclerosis and a history of viral infection, even when this has occurred in early childhood, the lack of a silent period or an extremely short silent period between the acute seizures at the time of the infection, and the onset of a chronic epilepsy is a red flag which must be paid attention to. This holds true for epilepsy after VE, as well as for epilepsy after BM (Lancman and Morris, 1996; Lee et al., 1997). In a study from Melbourne, Australia, (O’Brien et al., 2002), 39 consecutive patients with anterior temporal lobectomy and a remote history of meningitis or encephalitis were compared with matched controls without a history of cerebral infection. All patients had non-lesional temporal lobe epilepsy and were followed for at least a year after surgery. The authors concluded that the age at cerebral infection, but not the type of infection, is predictive of postsurgical outcome independent of the finding of mesial temporal sclerosis on preoperative MRI.
Patients presenting with a history of “encephalitis” later during childhood, a shorter “silent period” between the acute infection and the onset of epilepsy, and a combination of temporal and extratemporal lesions are rarely good candidates for epilepsy surgery, with the exception of those with unihemispheric damage so severe as to justify a hemispherectomy/hemispherotomy. Patients with acute encephalitis in early childhood, but without a “silent period” prior to epilepsy onset, are also included in this subgroup. Characteristic EEG features are a pathological diffuse or widespread theta/delta activity and slow background, indicating a diffuse or multiregional affection of the brain. Subtle or diffuse MRI findings (e.g. moderate atrophy and enlargement of the ventricles) may be encountered as a result of the inflammatory process. Hippocampal atrophy in some of these patients should not be appreciated as an indicator of a more favourable postsurgical outcome. Of higher predictive value, besides the absence of a “silent period”, are seizure semiology and EEG findings. Anatomo-electroclinical correlations are hard to obtain in these situations and results with respect to postoperative seizure outcome are often disappointing, even after invasive recordings (see review ofSellner and Trinka, 2012). Of 5/8 patients with later acute infections in a Korean cohort (Lee et al., 1997), one had an Engel II and two each had an Engel III and Engel IV outcome. In the Montreal Neurological Institute series from Canada (Trinka et al., 2000), the vast majority of patients (n=13) with an acute encephalitis during adolescence or adulthood had an unfavourable postsurgical outcome, with the exception of three patients with an Engel II outcome. In a study from the Cleveland Clinic, USA (Lancman and Morris, 1996), younger age at cerebral infection and longer latency between the acute infection and the onset of epilepsy were predictive for unilateral MTLE, whereas later infection and shorter latency between infection and epilepsy were predictive for bilateral MTLE. Development of neocortical epilepsy was associated with shorter latency between infection and epilepsy, and younger age at infection. In this study, the average age at acute encephalitis or meningitis was five years in patients with unilateral MTL or neocortical epilepsies and 14 years in patients with bilateral MTLE (as confirmed by depth electrodes, with no surgery offered thereafter). In a more recent report from Barcelona, Spain (Donaire et al., 2007), epilepsy surgery was carried out in 4/9 patients with epilepsy after VE; all four were young at acute infection. The two patients with longer latencies became completely seizure-free; the two patients with shorter latencies had rare auras at follow-up. Based on the results of the presurgical evaluation, no surgery was offered to the five patients who had suffered encephalitis in adulthood.
Epilepsy after bacterial meningitis
Morbidity and mortality after bacterial meningitis
The lack of antibiotics in some cases and their delayed administration in others may account for the high mortality of bacterial meningitis (BM) in several African (Pelkonen et al., 2009; Ramakrishnan et al., 2009) and other developing countries (Al Khorasani and Banajeh, 2006; Singhi, 2011). Further predictors of a severe course are:
- –grave initial presentation, e.g. coma, impaired consciousness, low body temperature, and circulatory failure;
- –age <12 months;
- –male gender;
- –and prolonged seizures (Grimwood et al., 2000; Chandran et al., 2011; Bargui et al., 2012; Namani et al., 2013; Lucas et al., 2016).
Pathological alterations of the meninges that spare the underlying cortex do not necessarily provoke seizures. Thus, seizures during the acute phase of meningitis may indicate that the affected person is suffering from a meningoencephalitis. Moreover, seizures may be attributed to a decrease in seizure threshold by fever. Around 30-50% of children who survive acute meningitis suffer long-term sequelae, although these more often comprise hearing loss or cognitive problems than seizures. About one third of children with BM treated at a tertiary centre in Northern India had neurological sequelae after 12 months of follow-up; only 9% had seizures (Singhi et al., 2007). Numerous studies have aimed to identify the risk factors for an adverse outcome following bacterial meningitis (Grimwood et al., 1996; Pagliano et al., 2007; Vasilopoulou et al., 2011; Bargui et al., 2012; Namani et al., 2013). However, the risk factors for epilepsy after BM have not been analysed separately from those for other neurological sequelae in any of these studies.
Risk factors for epilepsy after bacterial meningitis
Overall, epilepsy after BM is considerably more infrequent than epilepsy after VE. In a meta-analysis from 1993, including 4,920 children with a history of meningitis from 19 studies published since 1955, 4% of patients had seizures at follow-up (Baraff et al., 1993). The rate of epilepsy after BM depends on the acute course (complicated vs. uncomplicated) and the length of follow-up. In a 12-year follow-up study from Australia, considering children and adolescents with early childhood meningitis, 3/49 (6%) with complicated and only 1/60 (2%) with uncomplicated acute meningitis had epilepsy after BM. At short-term follow-up (Namani et al., 2013), epilepsy after BM rates were 0% at two weeks and 1% at three months. In a population study from England and Wales with a five-year follow-up period (Bedford et al., 2001), a single-centre study from France with a medium follow-up of 10 years (Bargui et al., 2012), and a “long-term follow-up” monocentric study from Kosovo (Namani et al., 2013), epilepsy after BM rates were 7%, 9%, and 9%, respectively. “Long-term follow-up“ studies usually lack the duration necessary to include adults with the syndrome of MTLE after acute bacterial meningitis in early childhood. The duration of a “silent period” after the insult to the mesial structures in early childhood can extend to three decades (table 2). However, the inclusion of these adult cases is not expected to increase the rate of epilepsy after BM. Temporal lobe epilepsies with an onset of chronic seizures without a “silent period” of at least six months after the acute infection do not correspond to pure MTLE. Interestingly, seizures during the acute phase of meningitis in the neonatal period do not cause typical hippocampal sclerosis. This may be attributed to the reduced vulnerability of the hippocampus in neonates, due to the immature expression of excitatory synapses at this developmental stage (discussion in Holthausen, 1994).
Surgery for epilepsy caused by bacterial meningitis
In a large surgical series from the Freiburg Epilepsy Centre, Germany, from 1999 to 2015 (Ramantani et al., 2013a, 2013b, 2013c, 2017), only nine of 369 children underwent surgery for drug-resistant epilepsy after BM. Cerebral infections leading to brain scarring and subsequent refractory epilepsy occurred within the first year of life in all but one patient. In most cases, the MRI-visible cortical damage was extensive, including three cases with hemispheric and one with bilateral pathology. The rate of neurological deficit matched the extent of brain damage; five patients had a hemiparesis, and four had a visual field deficit. All patients were drug-resistant to five or more AEDs (mean: eight AEDs), much higher than for any other aetiology (Ramantani et al., 2014) within the same large surgical series. This was in line with an overall very long latency of epilepsy onset to surgery (mean: 10 years). A total of 3/9 patients underwent functional hemispherectomy and another three underwent multilobectomies: temporo-parieto-occipital, fronto-temporal, and temporo-occipital, in one case each. The remaining three patients underwent anterior temporal lobectomy with amygdalohippocampectomy. Interestingly, three of these patients had a positive histopathology for FCD; two FCD 1b and one FCD 2a. Hippocampal sclerosis was shown in 7/9 patients. After a mean follow-up period of six years, 4/9 had an Engel I outcome, two had Engel II, and three had Engel III. It should be noted that in all five cases with seizure recurrence, this occurred within the first weeks after surgery.
Overall, two main types of epilepsy may develop after BM, similar to those following viral infection: (1) MTLE and (2) neocortical, predominantly extratemporal epilepsy.
Most patients with drug-resistant epilepsy after BM, who undergo epilepsy surgery, suffer from MTLE with hippocampal atrophy (table 2), associated with meningitis in infancy or early childhood (Marks et al., 1992; Donaire et al., 2007). Patients with a history of meningitis in early childhood, mesial temporal sclerosis on MRI, and an epilepsy course concordant with the syndrome of MTLE (“silent period”!) are excellent surgical candidates (Lancman and Morris, 1996; Lee et al., 1997; Trinka et al., 2000; Donaire et al., 2007; Sellner and Trinka, 2013). It is postulated that febrile seizures during the acute phase of meningitis in early childhood cause hippocampal atrophy, whereas seizures during neonatal meningitis do not cause this type of damage. Bilateral hippocampal atrophy in epilepsy after BM beyond the neonatal period, without marked damage in other parts of the brain, is highly unlikely to occur. The six-case series with hippocampal atrophy after meningitis from Barcelona, Spain (Donaire et al., 2007), may give a different impression. Two patients in this study were offered temporal lobectomies and one had a functional hemispherectomy; all three became seizure-free. The other three were excluded from surgery because of bilateral memory deficits, bilateral sharp waves, and/or “extratemporal” semiology. With a single exception, none of these patients had meningitis in early childhood (in contrast to the patients in the other three publications listed in table 2), whereas all had a prolonged epilepsy course. The higher rate of bilateral temporal pathology after a cerebral infection later in life has been previously reported (Lancman and Morris, 1996). In this study, in that of Marks et al. (1992), as well as the studies listed in table 2, successfully operated patients with epilepsy after BM and unilateral MTLE had meningitis in early childhood.
The reported high rates of invasive recordings in studies of epilepsy surgery in patients with epilepsy after BM (Sellner and Trinka, 2012) may reflect a more cautious approach in patients with MTLE following bacterial meningitis. However, these rates may also be attributed to the fact that some of these publications are somewhat older, representing an era of more generous indication for invasive recordings in MTLE than nowadays. Experienced epileptologists should be able to classify an epilepsy post BM correctly as “pure MTLE”, “temporal lobe epilepsy plus” (Kahane et al., 2015; Barba et al., 2016), a neocortical temporal lobe epilepsy, or an epilepsy linked to “dual pathology”. Of particular value for the diagnosis of MTLE after BM is the history of a “silent period” after the acute seizures occurring during meningitis in early childhood. Of concern is the long duration of epilepsy to surgery in all four reports dealing with meningitis in early childhood and the onset of epilepsy at school age (latency between 21 and 26 years; table 2), which is considerably longer than for other MTLE patients. The benefits of epilepsy surgery for patients with epilepsy after BM in early childhood presenting with symptoms, signs and MRI findings (hippocampal atrophy) concordant with the diagnosis of MTLE are still not as widely known as they deserve to be. In the only report dealing exclusively with epilepsy after BM during the first 4 years of life, 12/13 underwent an anterior temporal lobectomy, resulting in seizure freedom in all cases (Davies et al., 1996).
Neocortical epilepsies after BM are focal, multifocal, or even hemispheric, and the presumed epileptogenic lesions are MRI-visible. These correspond to neocortical scars as a result of a meningoencephalitis with a severe course or as a result of meningitis with late or ineffective antibiotic treatment. With the exception of patients with dramatic, widespread bi-hemispheric brain damage, a correlation of seizure semiology, interictal and ictal EEG findings with the localization of the MR-visible epileptogenic neocortical scars is feasible in most cases.
It should be noted that the extent of the epileptogenic zone in neocortical epilepsy after BM and its full or partial overlap with the extent of MRI-visible cortical scar(s) may differ considerably between postnatal meningitis and meningitis occurring later in life. Despite the lack of conclusive publications on this topic, there is some evidence that the epileptogenic zone in postnatal epilepsy after BM is larger than the visible cortical scar, due to “acquired cortical dysplasia” (Marín-Padilla, 1999). This observation is analogous to the patterns of cortical scarring encountered in pre-/perinatal stroke and to watershed lesions after hypoxic-ischaemic events in mature neonates. This consideration should be kept in mind when discussing the indication of invasive recordings in patients with neocortical epilepsy after BM. Further analogies to the two other aetiologies mentioned above are evident in the presence of widespread white matter damage in some cases and, possibly, in the additional presence of benign focal epileptiform discharges of childhood. If clinicians fail to appreciate these interictal discharges as benign, a child with refractory epilepsy after BM might be unjustifiably rejected for epilepsy surgery.
In some cases, brain damage due to the acute meningitis itself or due to the prolonged hemiconvulsions during the acute illness is so extensive as to consider hemispherectomy/hemispherotomy. Children with refractory epilepsy and contralateral hemiparesis related to hemispheric post-infectious scarring may profit from a hemispheric resection or disconnection. In a recent hemispherotomy cohort (Ramantani et al., 2013c), only one of 52 children and adolescents had a porencephalic cyst due to meningitis at the age of 4 years.
Epilepsy after neurocysticercosis
Neurocysticercosis (NCC) is most prevalent in Middle and South America, sub-Saharan regions of Africa, South-East Asia, particularly areas of China, and northern India. However, there are huge regional variations (Winkler, 2012), not only from continent to continent, but also from region to region in a given country, from rural areas to cities in the same province, from regions with a predominantly Muslim population to neighbouring areas with a different religion or prevalent ethnic group. NCC is practically non-existent in the province of Kerala that has a very high rate of literacy, but was the underlying aetiology of symptomatic focal epilepsy in 48% of 558 children from another province in south India (Murthy and Yangala, 2000). It should be noted that young children are less often and less severely affected in comparison to adults.
Due to the increased streams of migration from countries in which NCC is endemic to non-endemic countries, it is likely that physicians in more developed parts of the world will see mores patients with this disease in the near future (Serpa and White, 2012). Despite the availability of NCC treatment recommendations, NCC patients may not be correctly diagnosed and treated in the developed world. This is due to the limited exposure of physicians in the developed world to this disease (Garcia and Del Brutto, 2005; Nash et al., 2006; Singhi, 2011; Del Brutto, 2012a; Del Brutto and García, 2012). Nevertheless, according to a recent review (Del Brutto, 2012b), the risk of acquiring the disease for people travelling between countries to endemic areas is low, though it may increase for long-term stays.
The gold standard in NCC diagnostics is neuroimaging. MRI is superior to CT (Singhi, 2011; Verma and Lalla, 2012), but MRI is far less available than CT in developing countries (which, however, is sufficient in most cases). Patients with NCC from the Indian subcontinent present more often with single lesions, whereas those from Africa, Middle- and South America present more often with multiple lesions (Winkler, 2012). There are absolute, major, minor and epidemiological criteria for the diagnosis of NCC (Del Brutto et al., 2001; Del Brutto, 2012a). The main differential diagnoses are metastases, toxoplasmosis, and tubercula in the brain. In the diagnostic workup, diffusion weighted images and MRI-spectroscopy may be useful (Del Brutto et al., 1996; Carpio, 1998).
The majority of NCC cases are asymptomatic (Sanchez et al., 1999; Singhi et al., 2000; Fleury et al., 2003; Montano et al., 2005; de Almeida and Torres, 2011; Prasad et al., 2011). However, epileptic seizures are the main neurological manifestation, present in over 90% of symptomatic NCC (Monteiro et al., 1992; Singhi et al., 2000; Medina et al., 2005). Seizures in NCC, particularly new-onset seizures, seem to occur more often during the active and transitional stages than during the degenerative stage of NCC (Carpio, 1998), although regional differences may again be noted (Singhi, 2011).
Incidence and prevalence of epilepsy after neurocysticercosis
NCC resulting from infection with the larvae of the porcine tapeworm Taenia solium is the most frequent cause of new-onset epilepsies in many parts of the world, mainly in developing countries (Relationship between epilepsy and tropical diseases, 1994; Román et al., 2000; Garcia and Del Brutto, 2005). NCC is reportedly the underlying cause in up to 30% of epilepsies in the developing world (Burneo et al., 2009). Particularly grim figures regarding the burden of NCC-associated chronic epilepsy in sub-Saharan countries are outlined in a recent review (Winkler, 2012). These figures are high because, among other reasons, many affected persons in resource-poor countries do not take AEDs or do not take them regularly. Extremely puzzling is the considerable variability in chronic epilepsy prevalence in countries where NCC is endemic. NCC was the cause of new-onset epilepsy in 50% of affected persons in a population-based study from a rural area in Peru (Villarán et al., 2009), and the cause of symptomatic epilepsy in only 12/346 (4%) patients in a report from West-China. Figures of around 30%, 37%, and 37% are reported in publications from Peru and Honduras (Gaffo et al., 2004; Medina et al., 2005; Montano et al., 2005, respectively). A total of 48% of children with epilepsy seen at the university hospital in Hyderabad, India, had epilepsies associated with NCC (Murthy and Yangala, 2000).
Drug-resistance in NCC
Most publications on NCC-related epilepsy support that seizures are controlled by AEDs more successfully than for most other aetiologies. This holds true especially for some recently reported paediatric cases from India. Features contributing to this favourable course are the presence of predominantly non-calcified lesions in children (15% calcified lesions vs. 55% in adults), as well as the tendency of lesions to disappear during follow-up in a quarter to a third of cases (Gadgil and Udani, 2011). The recurrence rate after AED withdrawal is higher in patients with multiple and calcified lesions (Singhi, 2011). However, it is difficult to get a clear picture regarding the prevalence of drug-resistant epilepsies. Only 8/512 (2%) patients with drug-resistant epilepsies seen at the outpatient clinic in Ribeirao Preto in Brazil had “isolated neurocysticercosis” due to calcified lesions. Calcified lesions were found in 27% of patients with MTLE. However, according to the physiciańs view in this renowned epilepsy centre, these lesions were unrelated to the MTLE.
Two major issues remain unanswered regarding epilepsy after NCC. It is still unclear whether, in patients with NCC, hippocampal sclerosis results from recurrent seizure activity from a local or distant focus or chronic recurrent inflammation (Singla et al., 2007; Del Brutto et al., 2016). In either case, hippocampal sclerosis may become the pathological substrate of subsequent MTLE. The role of calcified NCC lesions in epilepsy pathogenesis and their inter-relations with co-existing hippocampal sclerosis are yet to be clarified (Singh et al., 2000; Nash et al., 2004; Velasco et al., 2006; Singla et al., 2007; Bianchin et al., 2013; de Oliveira Taveira et al., 2015). Four different hypotheses have been considered:
- –coincidence, since both entities are relatively common in endemic areas;
- –calcified NCC lesions and MTLE related to hippocampal sclerosis are associated, but not causally related;
- –NCC may directly cause MTLE related to hippocampal sclerosis by inflammation and structural damage to the hippocampi or cortex by the acute phase of cysticerci located in the vicinity -apparently not applicable for non-temporal NCC;
- –and NCC is rather a trigger than the direct cause of the epileptogenic process leading to MTLE.
Surgery for epilepsy after neurocysticercosis
For a condition so often linked to chronic epilepsy, one would expect that a significant proportion of patients would undergo epilepsy surgery, especially since there are excellent epilepsy surgery centres in countries where NCC is endemic, such as China, Brazil, India, and South Africa. Nevertheless, the numbers of operated patients with refractory epilepsy caused by NCC are very low, whereas most of them have MTLE (Leite et al., 2000; da Gama et al., 2005; Chandra et al., 2010; Bianchin et al., 2013; Rathore et al., 2013; Meguins et al., 2015).
In a study from the epilepsy centres in Ribeirao Preto and Campinas, Brazil, no clinical or electroencephalographic differences were found between patients with MTLE and those with MTLE linked to NCC-related calcified MRI lesions (da Gama et al., 2005; Leite et al., 2000). Furthermore, these two groups did not differ in surgical outcome. Therefore, it was concluded that the calcified lesions in these patients were coincidental findings in a population where NCC is endemic and where most affected persons are asymptomatic. This conclusion has been recently challenged in a study with favourable postsurgical outcomes linked to the resection of calcified lesions, located within the temporal lobe, in addition to the resection of the mesial temporal structures (Rathore et al., 2013). Furthermore, although there was no difference regarding Engel I outcomes, more patients with MTLE alone had Engel Ia outcomes compared to patients with MTLE plus NCC. An excellent discussion about the issue of whether NCC-related calcified lesions are causative or not in MTLE can be found in a recent review (Singh and Chowdhary, 2014).
Reports of extratemporal epilepsy surgery in patients with NCC and epilepsy are exceedingly rare. In a study from India, 4/5 patients were seizure-free after extratemporal epilepsy surgery (Rathore et al., 2013). In a case report from Malaysia, the resection of a cysticercotic granuloma within the left frontal lobe led to seizure freedom (Hasan et al., 2011). The paucity of data on extratemporal surgery for NCC-related epilepsy may be attributed to the fact that even experienced epilepsy centres may hesitate to proceed to surgery when multiple lesions are present. Another reason may be that epilepsies caused by singular extratemporal NCC-related calcified lesions usually respond to AEDs. Several authors have discussed the pathophysiological mechanisms responsible for rendering NCC-related calcified lesions epileptogenic. One possible mechanism is immunoreaction by the host leading to a (subtle) inflammatory “perifocal reaction”. Perfusion MRI may help to differentiate epileptogenic from non-epileptogenic calcified lesions (Gupta et al., 2012).
Surgery for epilepsy caused by rare infectious agents
On very rare occasions, patients with cerebral tuberculomas, schistosomiasis, and other rare infectious agents may undergo epilepsy surgery.
Tuberculosis
In a five-case epilepsy surgery series from Bangladesh (Chowdhury et al., 2010), one of the patients suffered from a tuberculoma. The patient presented typical mesial temporal seizures with increasing frequency for over almost a year without any neurological deficit or deterioration of general condition or cognitive functions. MRI showed a hyperintense lesion in the anterior/mesial temporal lobe, initially attributed to meningioma or ependymoma. The patient underwent standard anterior temporal lobectomy with amygdalohippocampectomy. Histopathology revealed brain tuberculosis. At the last follow-up visit, at five months, the patient was seizure-free without taking AEDs. Tuberculosis is not rare in Bangladesh, but its presentation with MTLE without any systemic symptoms and signs is unusual. A study on epilepsy surgery for post-infectious encephalitis from New Delhi, India (Chandra et al., 2010) included three patients with tuberculosis, as well as a case of gliotic scars and multiloculated hydrocephalus following tuberculosis meningitis in the first year of life. This patient with the extensive tuberculosis-related brain damage underwent hemispherotomy. The remaining three patients underwent a resection of the mesial temporal structures, a neocortical temporal resection, and a frontal resection. At the last follow-up visit, at over a year after surgery, all four patients were seizure-free.
Schistosomiasis
Schistosomiasis is the most common trematode infection, widely distributed in Asia, Africa, and Latin America. Schistosomal granuloma results from a host-to-pathogen interaction that triggers an immune response and manifests similarly to a brain tumour. In a large surgical series from the Wuhan Tongji Hospital, China in 1955-2004 (Lei et al., 2008), 250 patients with seizures as the initial symptom of cerebral granulomas caused by Schistosoma japonicum and drug-resistant epilepsy later on underwent surgery 2-12 years after epilepsy onset. After 4-5 years of follow-up, four patients died as a result of the schistosomal liver cirrhosis and ten died of natural causes; of the remaining 196 cases, 180 (92%) were seizure-free. All patients in this series presented chronic schistosomiasis with hepatosplenomegaly, ascites, and esophagogastric varices in more than half of the cases.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Disclosures
None of the authors have any conflict of interest to declare.