Epileptic Disorders
MENUEncephalopathy related to Status Epilepticus during slow Sleep: a link with sleep homeostasis? Volume 21, supplement 1, June 2019

Sleep is beneficial for cognition by actively participating in learning, language acquisition and memory consolidation. A wealth of experimental data in healthy adults and children have demonstrated the positive effects of post-training sleep in the consolidation of recently learned information (Stickgold et al., 2000; Walker et al., 2002; Fenn et al., 2003; Diekelmann and Born, 2010; Schreiner and Rasch, 2017). These works imply that chronic sleep disturbances in the critical period of brain maturation may have adverse effects on learning and development.
In recent years, a growing body of evidence has suggested that a sleep disruption due to epileptic activity may play a role in the cognitive impairment observed in children suffering from epilepsy (Holmes and Lenck-Santini, 2006; Parisi et al., 2010; Chan et al., 2011; Urbain et al., 2013). Several studies have demonstrated in benign focal epilepsies of childhood as well as in epileptic encephalopathies an association between cognitive and behavioural dysfunctions and the amount of epileptic activity during NREM sleep, supporting the hypothesis that epileptic discharges during NREM sleep can exert negative effects on cognitive abilities (Tassinari and Rubboli, 2006; Massa et al., 2011; Ebus et al., 2011; Van Bogaert et al., 2012; Filippini et al., 2013). Further support to this hypothesis is provided by data showing that improvement of cognitive abilities is strongly correlated with reduction or disappearance of epileptiform discharges during sleep even though normalization of the EEG does not appear to be the only predictive factor (Soprano et al., 1994). Yet, the pathophysiological mechanisms linking the appearance of neuropsychological deficits and epileptic activity during sleep remain largely speculative. Here we review some recent data on a peculiar childhood epileptic syndrome that can represent a model for the investigation of the complex and reciprocal interactions among epileptic activity, sleep physiology and cognitive functions in the developmental age, i.e. the “Encephalopathy related to Status Epilepticus during slow Sleep” (ESES) and we discuss how these findings may provide some clues for the understanding of the links between cognitive impairment, sleep disruption, and epileptic activity during NREM sleep.
EEG and cognition in ESES
ESES is an age-dependent self-limited epileptic condition characterized by:
- –onset in infancy or childhood (with a peak around the age of 4-5 years);
- –heterogeneous types of epileptic seizures;
- –a typical EEG pattern characterized by continuous or subcontinuous epileptic activity during non REM (NREM) sleep that can last for several months or years;
- –variable neuropsychological regression (consisting of IQ decrease, reduction of language), disturbances of behavior (such as development of autistic features, psychotic states), and motor impairment occurring in conjunction with the appearance of the abnormal sleep EEG pattern (Tassinari et al., 2000).
In spite of the favourable long-term prognosis of the epilepsy and of the normalization of the EEG, cognitive deficits and behavioural disorders may persist for life.
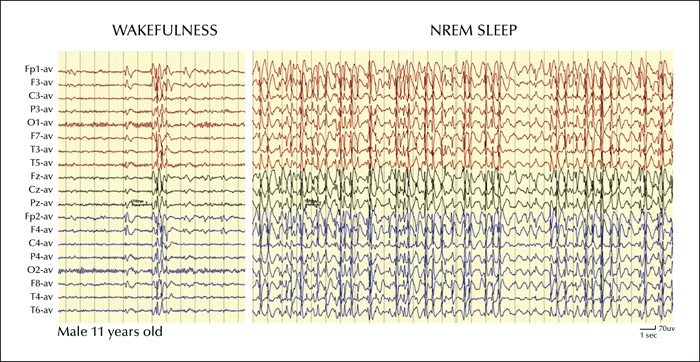
A striking feature of ESES is the extreme amount of epileptic discharges occurring during sleep, up to a spike-wave index (SWI) of 85-100% of the time spent in NREM sleep (figure 1). This feature led to the eponym of “Electrical Status Epilepticus during Sleep” in the original description by Patry, Lyagoubi and Tassinari (Patry et al., 1971). A few years later, Tassinari et al. (1977) reporting an additional series of patients, proposed that the “status epilepticus during sleep” (i.e., SES) was responsible for an encephalopathy characterized primarily by cognitive and behavioural dysfunctions and named it “Encephalopathy related to Status Epilepticus during slow Sleep” (i.e. ESES). The SES is thought to have a focal cortical origin that acts as a trigger for the spread of epileptic abnormalities through a secondary bilateral synchrony mechanism (Blume and Pillay, 1985; Kobayashi et al., 1994). A focal origin of the epileptic discharges is suggested by the focal nature of the main seizure type, by the observation of focal EEG activity in wakefulness or REM sleep, by intracerebral EEG recordings (Solomon et al., 1993), by the spike phase reversal and interhemispheric peak latencies over unilateral regions (Morikawa et al., 1995), and by focal metabolic abnormalities demonstrated by functional imaging studies (PET, SPECT, fMRI studies) (Hirsch et al., 1990; Mouridsen et al., 1993; Maquet et al., 1995; De Tiège et al., 2009; Siniatchkin et al., 2010). The focal epileptic activity would then engage thalamo-cortical networks underlying the slow oscillations of sleep (Destexhe and Sejnowski, 2001) and enhance their synchrony, promoting their evolution into spike-wave seizures (Amzica and Steriade, 2000) (see also Gibbs et al., p. S54-61). Changes in thalamic firing levels can also switch the cortical dynamics from a desynchronized to a synchronized state and contribute to the synchronization of spike-wave discharges in the cortex (Hirata and Castro-Alamancos, 2010; Liu et al., 2015). These mechanisms may play a role also in the progression and possibly in the maintenance of SES. A role of the thalamus is also supported by evidence of early thalamic injuries in children with ESES (Guzzetta et al., 2005; Sánchez Fernández et al., 2012).
Besides the peculiar sleep EEG pattern, the most intriguing aspect of ESES is the relationship between the onset of SES and the appearance of an encephalopathy accompanied by prominent cognitive and behavioural disturbances, which may persist permanently, or only partially recover, after the resolution of SES and disappearance of seizures (Tassinari et al., 2000, 2012; Seegmüller et al., 2012; Caraballo et al., 2013, see also Arzimanoglou and Cross, p.S71-5, and Dorris et al., p.S88-96). The onset of neuropsychological symptoms follows closely the onset of status epilepticus, and the degree and type of cognitive/behavioural dysfunction seem to depend on the duration and localization of the focal epileptic activity (Tassinari and Rubboli, 2006; Tassinari et al., 2012). These observations have led to the hypothesis that epileptic discharges during sleep may disrupt cognitive and/or motor functions, even when they remain subclinical. Indeed, such epileptic activity (Patry et al., 1971), while originally considered “subclinical” because of the lack of overt electro-clinical correlations during sleep, becomes undoubtedly clinical albeit belatedly, due to its effects on cognition and behaviour during wakefulness. These concepts, proposed by Tassinari et al. in 1977, have been assimilated into the recent definition of “epileptic encephalopathy” by the International League Against Epilepsy (ILAE) Classification Task Force. This term encompasses not only the conditions with frequent seizures but also those with abundant “interictal” epileptiform abnormalities in which epileptic activities “per se” exert an effect in the development of severe cognitive and behavioural impairments “above and beyond what might be expected from the underlying pathology alone” (Berg et al., 2010).
Sleep homeostasis and cognition
What are the pathophysiological mechanisms by which prolonged epileptic activity during sleep leads to the development of the cognitive impairment and behavioural derangement? An intriguing possibility is that the negative effects of ESES may be linked to an excessive engagement of the process of synaptic homeostasis thought to occur during normal sleep (Tassinari and Rubboli, 2006).
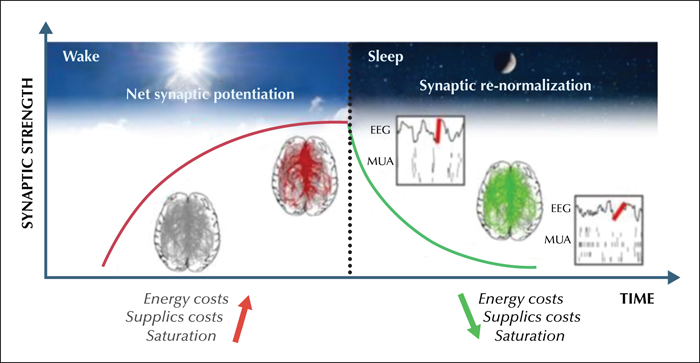
Sleep is a homeostatic process: the longer we stay awake, the more intensely we need to sleep, and only sleep can lead to brain restoration after wakefulness (Borbély and Achermann, 2005). Sleep need is reflected in EEG slow wave activity (SWA, 1-4,5 Hz) during NREM sleep in all mammalian species investigated so far: SWA increases exponentially as a function of prior wakefulness and decreases exponentially during sleep (Tobler, 2000; Borbély and Achermann, 2005). In recent years, a number of studies have shown that sleep homeostasis is closely related to cortical plasticity processes (Tononi and Cirelli, 2014). Specifically, wakefulness, which is typically associated with learning and memory formation, leads to a net increase in synaptic strength and to cellular stress. By contrast, sleep promotes net synaptic depression and renormalization, memory consolidation, and cellular recovery (Vyazovskiy et al., 2008; Maret et al., 2011; Bushey et al., 2011; Tononi and Cirelli, 2014; de Vivo et al., 2017; Diering et al., 2017). Furthermore, both human and animal studies have demonstrated a link between changes in synaptic strength and sleep SWA. A net increase in synaptic strength during wakefulness leads to stronger coupling and therefore increased synchrony among neurons, which is reflected in slow waves of larger amplitude that are steeper during sleep (Esser et al., 2007; Vyazovskiy et al., 2009). On the other hand, slow waves occurring during sleep are thought to promote a progressive down-selection of synapses, which in turn reduces the amplitude of slow waves and, under normal condition, reaches a set point at which SWA is sufficiently low and down-selection stops (figure 2). Importantly, the homeostatic regulation of sleep slow waves also has a local component. Learning tasks that produce local strengthening of connections are followed by a local increase in sleep slow waves (Huber et al., 2004), whereas interventions that lead to non-physiological synaptic depression such as arm immobilization or low frequency TMS are followed by a local decrease in slow wave activity (Huber et al., 2006) (reviewed in Tononi and Cirelli, 2014).
Synaptic plasticity and sleep homeostasis
The link between synaptic plasticity and sleep homeostasis is likely to be especially important during development. The architecture of synaptic connections in the cerebral cortex is wired and optimized throughout childhood and adolescence (Rakic et al., 1994). The balancing of synaptic strength across sleep and waking may thus be essential for cortical maturation and thus normal cortical functioning. Intriguingly, sleep slow wave activity peaks during childhood and decreases progressively in the course of adolescence in a way that appears correlated with the occurrence of synaptic refinement in cortical circuits (Huttenlocher, 1979; Feinberg, 1982; Jha et al., 2005; Campbell and Feinberg, 2009; Nelson et al., 2013; Olini et al., 2013; de Vivo et al., 2014; Hoel et al., 2016). Moreover, the topographic distribution of SWA during cortical maturation highlights the developmental changes in cortical plasticity: SWA is highest over posterior regions during early childhood and then shows a spatial shift to anterior regions, reaching the frontal cortex in late adolescence (Kurth et al., 2010). Critically, the maturation of specific skills is predicted by the topographical distribution of SWA, the more frontal the topography of SWA, the better adolescents perform in tasks known to be frontally-mediated (Kurth et al., 2012). Thus, both human and animal studies show a close correspondence between the maturation of the cortex, SWA and behaviour.
Impaired synaptic homeostasis in ESES
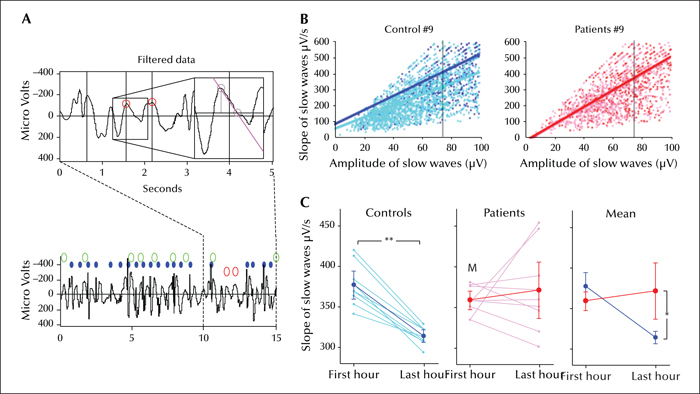
The occurrence of massive spike wave activity during slow wave sleep every night in children with ESES would be expected to interfere with sleep homeostasis. A sensitive indicator of changes in synaptic strength and associated sleep homeostasis is the slope of sleep slow waves (Esser et al., 2007; Riedner et al., 2007; Vyazovskiy et al., 2007). In healthy control subjects (adults, adolescents and children), the slope of slow waves decreases across sleep (Riedner et al., 2007; Kurth et al., 2010). Children suffering from ESES do not show such a decrease, as if the homeostatic recovery of sleep were impaired (Bolsterli et al., 2011) (figure 3). A follow-up study showed that the impairment was most severe at the epileptic focus and that this impairment was positively correlated with the SWI (Bolsterli Heinzle et al., 2014). Moreover, while healthy children showed an increased recall of learned word pairs after a night of sleep, children with ESES showed instead a decrease (Urbain et al., 2011). In one child, hydrocortisone treatment normalized both the sleep EEG and overnight performance (another child whose EEG was only partially improved showed no improvement), suggesting that the impairment of sleep homeostasis may be responsible for the lack of memory consolidation. Finally, Boelsterli et al. (2017) have recently shown in children with idiopathic ESES that cognitive/behavioural outcome might correlate with the degree of impairment of the slope of the sleep slow waves. Indeed, complete cognitive/behavioral recovery after ESES was observed in those children in whom the overnight decline of the slope of sleep slow waves during ESES active phase was partially preserved; on the contrary, lack of decline of the slope of sleep slow waves was associated with persistent neuropsychological deficits after ESES resolution. Interestingly, reduced overnight slope decline of sleep slow waves has been shown to be related to poorer learning during wakefulness also in adult patients with focal epilepsy (Boly et al., 2017).
Spikes, synaptic homeostasis and cognition
If normal sleep plays a fundamental role in synaptic renormalization, it is natural to ask whether epileptic activity during NREM sleep may interfere with the homeostatic renormalization of synapses and produce instead abnormal synaptic potentiation. Such abnormal plasticity, recurring nightly for months and years, might eventually lead to a severe impairment of the cognitive functions and behaviour mediated by the affected brain areas (Buzsaki, 1998; Tassinari and Rubboli, 2006; Romcy-Pereira et al., 2009). In animal models, epileptic discharges occurring during wakefulness lead to long-term synaptic potentiation (LTP) (Buzsáki, 1989; Leite et al., 2005; Romcy-Pereira et al., 2009) by hijacking the mechanisms of learning in a maladaptive manner, unrelated to environmental inputs. It is likely that epileptic discharges occurring during sleep also induce plastic changes, although it is unclear in which direction: potentiation, as is normally the case in wakefulness, or depression, as is normally the case in NREM sleep.
The SWA of NREM results from an ongoing alternation between a depolarized ‘up-state’, when neurons fire tonically, and a hyperpolarized ‘down-state’, when neurons stop firing for hundreds of milliseconds (Amzica and Steriade, 1998). Laminar recordings of local field potentials, and of multiple- and single-unit activities during intracerebral recordings in humans have shown that the ‘up-state’ of sleep slow waves is associated with multi-unit-activity bursts as well as high-frequency oscillations (HFO) (Csercsa et al., 2010; Nir et al., 2011). In cortical areas heavily involved in learning processes, SWA, slow wave slopes, and the synchrony of HFO during the “up-state” increase together. Intriguingly, a recent study in ESES has demonstrated the occurrence of exaggerated HFO related to epileptic spikes (Kobayashi et al., 2010). Such pathological HFO occurring systematically throughout NREM sleep are likely to produce major abnormalities in synaptic plasticity and impair the synaptic renormalization processes.
Such an impairment would be especially harmful during the critical period of cortical maturation in childhood and pre-adolescence, which is characterized by massive synaptogenesis, synaptic pruning, and the refinement of synaptic circuits (Huttenlocher, 1979; Huttenlocher and Dabholkar, 1997; Petanjek et al., 2011; Liu et al., 2012). In this critical period, plastic changes involve all cortical laminae and thalamo-cortical inputs (Issa, 2014) and local increases of learning-related SWA are strongest (Wilhelm et al., 2014). It is well-established that the maturation of cortical circuits during adolescence occurs in parallel with a progressive reduction of SWA (Buchmann et al., 2011; Campbell et al., 2011). Recently, computer simulations have shown that this developmental decline of SWA can track activity-dependent synaptic refinement that underlies the proper functioning of neural circuits (Hoel et al., 2016). In animal models, interfering with cortical activity during NREM sleep leads to a deterioration of previously acquired cortical adaptations (Frank et al., 2001). Altogether, these data suggest that a systematic impairment of synaptic renormalization during NREM sleep would lead to profound, possibly irreversible changes in cortical wiring, which may thus represent an unfortunate consequence of “subclinical” epileptic activity in the active phase of ESES. Such a model would provide an explanation for the loss of acquired functions, such as language, and for the persistence of cognitive and behavioural disturbances after the end of ESES. The report of prefrontal lobe growth during the active phase in a patient with ESES may also be a macroscopic anatomical consequence of impaired synaptic renormalization during sleep (Kanemura et al., 2009).
In summary, an impairment of synaptic renormalization due to epileptic activity during NREM sleep could explain many of the clinical features in children with ESES, and possibly in other forms of childhood epilepsy with a striking increment of paroxysmal abnormalities during NREM sleep or with altered slow wave activity (e.g. West syndrome patients; Fattinger et al., 2015). The clinical implication is that such “subclinical” abnormalities during critical developmental periods should be seriously considered, as they may alter cortical wiring and thereby disrupt cognitive and behavioural functions irreversibly. Future studies should explore whether in epileptic children with high SWI during sleep and absent or minor cognitive disturbances, the reduction of the slope of the slow waves during sleep is preserved or minimally affected, reflecting a preservation or minimal perturbance of synaptic renormalization. This realization calls for careful testing, including neuropsychological and behavioural tools to detect selective or subtle cognitive dysfunctions related to a highly focal ESES (Kuki et al., 2014), that coupled with sophisticated neuroimaging, and the use of novel EEG indices, in addition to SWI, such as the decline of slow wave slopes in NREM sleep, might be useful for a timely diagnosis and treatment, and might provide indirect information on the cortical networks underlying specific cognitive functions (Tassinari et al., 2015).
Disclosures
None of the authors have any conflict of interest to declare.