Hépato-Gastro & Oncologie Digestive
MENUMastocytosis: Gastrointestinal symptoms Volume 25, issue 8, Octobre 2018

Introduction
Les mastocytoses sont des maladies rares. On distingue classiquement les formes cutanées pures (90 %) et les formes systémiques (10 %). En 2006, en France 1 300 cas de cas de mastocytose ont été recensés par l’Association française pour les initiatives de recherche sur le mastocyte et les mastocytoses (AFIRMM) mais ce chiffre est probablement fortement sous-estimé. La mastocytose cutanée pure est une maladie essentiellement pédiatrique, au cours de laquelle les mastocytes envahissent uniquement la peau, qui disparaît le plus souvent lors de la puberté. Les mastocytoses systémiques (MS) sont au contraire des maladies chroniques essentiellement de l’adulte (âge moyen au diagnostic : 60 ans) où les mastocytes s’accumulent également dans d’autres organes (moelle, foie, rate, tube digestif, os…). Les manifestations cliniques sont variables, soit en rapport avec la libération de médiateurs mastocytaires, soit la conséquence d’une infiltration tumorale. Les mastocytoses au moins chez l’adulte peuvent être assimilées à des syndromes myéloprolifératifs. La mise en évidence d’une mutation du gène C-KIT, le plus souvent la D816V, chez la plupart des patients présentant une forme sporadique de mastocytose suggère un rôle essentiel de cette tyrosine kinase dans l’origine de la maladie et est maintenant une cible thérapeutique. La présence de mutations activatrices de C-KIT entraîne une autophosphorylation à l’origine de la différenciation, de la migration et de l’accumulation de mastocytes dans les différents tissus [1]. Les mastocytoses systémiques sont des maladies chroniques de l’adulte où les mastocytes s’accumulent également dans d’autres organes (moelle, foie, rate, tube digestif, os…)
La plupart des manifestations liées à la dégranulation mastocytaire sont paroxystiques, survenant spontanément ou déclenchées par divers facteurs ou stimuli. On citera les flushs, des réactions anaphylactoïdes, un prurit généralisé, des atteintes osseuses (dont la physiopathologie est mal connue), des manifestations respiratoires à type de dyspnée, voire de bronchospasme, d’hypersécrétion de mucus ou d’œdème pulmonaire, des manifestations digestives, qui sont parfois au premier plan. Des troubles neuropsychiatriques sont fréquemment observés (dépression, anxiété, troubles de la mémoire…). Enfin, la cystite interstitielle est un symptôme très fréquent mais peu connu qui se manifeste par une pollakiurie et est liée à la libération locale de médiateurs par les mastocytes infiltrant la muqueuse vésicale. La plupart des manifestations liées à la dégranulation mastocytaire sont paroxystiques, survenant spontanément ou déclenchées par divers facteurs ou stimuli
Les manifestations liées à l’infiltration tumorale comportent des symptômes cutanés (notamment à type d’urticaire pigmentaire), une infiltration médullaire (présente dans 90 % des mastocytoses systémiques), un syndrome tumoral avec hépatosplénomégalie, des manifestations digestives (gastro-intestinales, hépatobiliaires) et de rares atteintes cardiopulmonaires. Les manifestations liées à l’infiltration tumorale comportent des symptômes cutanés, une infiltration médullaire, un syndrome tumoral avec hépatosplénomégalie, des manifestations digestives (gastro-intestinales, hépatobiliaires) et de rares atteintes cardiopulmonaires
Le diagnostic formel de mastocytose s’établit sur des données histologiques mais dans certaines formes sans atteinte cutanée ou avec des manifestations systémiques, des examens complémentaires sont nécessaires à la fois pour confirmer le diagnostic, établir un pronostic et définir la meilleure stratégie thérapeutique. L’OMS a défini en 2001 et révisé en 2016 les critères diagnostiques de MS. Si un critère majeur et un critère mineur, ou trois critères mineurs sont présents chez un patient, on peut retenir le diagnostic de MS[3]. Le critère majeur est défini par la présence d’infiltrats denses (> 15 mastocytes par agrégat confirmés par l’immuno-histochimie ou les colorations spécifiques) et multifocaux de mastocytes dans une ou plusieurs biopsies d’organes en dehors de la peau (dans la plupart des cas la biopsie médullaire). Les critères mineurs comportent : présence de mastocytes atypiques de forme allongée ou de promastocytes dans la moelle (plus de 25 % des mastocytes) ; présence de la mutation D816V de C-KIT dans un ou plusieurs organes (surtout au niveau médullaire) ; coexpression du CD117 avec le CD2 et/ou le CD25 par les mastocytes médullaires, du sang ou d’autres organes ; élévation de la tryptase sérique au-dessus de 20 ng/mL (critère invalide s’il coexiste une hémopathie non mastocytaire).
Les mastocytoses constituent une entité très hétérogène faite de maladies différant aussi bien par leur présentation que leur pronostic et leur traitement. Plusieurs classifications ont été proposées, dont la plus récente, établie par l’OMS en 2016 [1].
Dans les formes systémiques indolentes ou cutanées pures, la maladie ne diminue pas l’espérance de vie des malades, mais elle reste associée à un handicap important, mal connu, lié à la libération des médiateurs mastocytaires et qui pose parfois des problèmes diagnostiques, quand l’infiltration mastocytaire n’est pas au premier plan, et thérapeutiques.
Les aspects cliniques, physiopathologiques et thérapeutiques des formes gastro-intestinales (excluant donc les atteintes hépatiques, biliaires et spléniques) sont discutés dans cette mise au point.
Symptômes gastro-intestinaux au cours de la mastocytose systémique
La fréquence rapportée des symptômes gastro-intestinaux au cours de la MS varie selon les études (tableau 1). Les premières grandes études [2, 3]rapportaient une fréquence de l’ordre de 20 % alors que la majorité des études ultérieures jusqu’à 1985 rapportaient une fréquence de l’ordre de 40-50 % [4-6]. Les études réalisées depuis 1985 rapportent une fréquence plus élevée, de 60 à 80 %, rendant les symptômes gastro-intestinaux aussi fréquents que le prurit chez ces patients [7-10]. Plusieurs facteurs peuvent contribuer à cette grande variabilité rapportée dans les séries : la variabilité dans la définition de la mastocytose, dans la définition des symptômes digestifs et de leur intensité, et dans le type de recueil de données (rétrospectif vs. prospectif).
Plusieurs études soulignent que les symptômes gastro-intestinaux chez les patients atteints de MS sont chroniques et sont une source majeure d’altération de la qualité de vie. Les symptômes gastro-intestinaux chez les patients atteints de mastocytoses systémiques sont chroniques et sont une source majeure d’altération de la qualité de vie
La douleur abdominale est le symptôme le plus fréquent, touchant approximativement un patient sur deux. La diarrhée est le second symptôme le plus fréquent (de l’ordre de 40 %), puis viennent les nausées et vomissements (30 %). La fréquence des hémorragies digestives, quand elle est rapportée, est de l’ordre de 10 %. Concernant la maladie ulcéreuse peptique, les données issues d’études anciennes sont soumises à caution car elles ne comportaient pas d’examen endoscopique le plus souvent. Dans les études plus récentes, la fréquence est de l’ordre de 25 %. La douleur abdominale est le symptôme le plus fréquent touchant approximativement un patient sur deux. La diarrhée est le second symptôme le plus fréquent (de l’ordre de 40 %), puis viennent les nausées et vomissements (30 %)
Les atteintes digestives liées à la mastocytose peuvent toucher la totalité du tractus digestif. Les atteintes œsophagiennes sont rarement mentionnées dans les cas de MS. Des symptômes ou lésions compatibles avec un reflux gastro-œsophagien (pyrosis, œsophagite, sténose œsophagienne peptique) ont été rapportés dans quelques cas [16]. Des cas de troubles moteurs de l’œsophage ont également été rapportés et semblent fréquents au cours de la MS, avec notamment des anomalies de fonction du sphincter inférieur de l’œsophage (diminution de pression de repos, absence de relaxation à la déglutition) [12, 13]. Dans la plupart des études, la majorité des patients atteints de MS ont une augmentation de la production d’histamine. L’histamine étant un stimulant bien connu de la sécrétion acide gastrique, la majorité des patients devrait avoir un certain degré d’hypersécrétion acide gastrique. Dans les différents cas rapportés, la sécrétion acide basale était très variable : soit élevée, soit normale, soit diminuée [5]. Dans la seule étude prospective évaluant ce paramètre au cours de la MS [7], la sécrétion acide basale était augmentée chez 6 des 16 patients étudiés (38 %). Cinq de ces six patients avaient d’ailleurs une endoscopie haute anormale. Dans cette même étude, un des patients avait une achlorhydrie complète, comme cela avait déjà observé dans quelques cas antérieurs [5]. On peut noter qu’alors que la totalité des patients de l’étude de Cherner ont une élévation de leur taux sérique d’histamine, plus de 50 % ont une sécrétion acide basale normale. Les possibles explications pour cette dissociation entre élévation du taux sérique d’histamine et l’hyperacidité gastrique sont (i) que l’histamine mesurée n’est pas active biologiquement, (ii) que l’hyper-histaminémie n’est pas suffisante pour stimuler les cellules pariétales gastriques, (iii) que les récepteurs H2 de l’histamine des cellules pariétales gastriques sont désensibilisés (des taux très élevés d’histamine sont donc nécessaire à leur activation), ou encore (iv) que le taux circulant d’histamine n’est pas le déterminant majeur de la sécrétion acide gastrique (la concentration muqueuse gastrique d’histamine importerai plus dans ce cas). La sécrétion acide maximale a été moins souvent étudiée chez les patients atteints de MS. Dans l’étude par Cherner, elle était mesurée chez 16 patients et était augmentée seulement dans 12 % des cas, alors que tous les patients avaient une hyperhistaminémie sérique [7].
Des lésions gastriques urticariennes ont été observées chez plusieurs patients. Des lésions de gastrite avec infiltrat inflammatoire de la muqueuse et de la sous-muqueuse et présence de mastocytes ont été observées chez plusieurs patients atteints de MS.
L’absorption intestinale est perturbée chez un certain nombre de patients atteints de MS [8, 9]. Une étude prospective portant sur 16 patients a mis en évidence un certain degré de malabsorption chez 5 d’entre eux (31 %) [7]. Néanmoins, lorsqu’il existe une stéatorrhée chez un patient atteint de MS, elle est minime ou modérée dans la très grande majorité des cas [5].
Des lésions histologiques de l’intestin grêle ont été observées chez de nombreux patients, mais la prévalence de ces lésions n’est pas connue étant donné l’absence d’étude systématique de séries de patients consécutifs. Dans de nombreuses études, les villosités intestinales sont souvent émoussées voir même parfois partiellement atrophiques [10, 14]. Quelques cas d’atrophie villositaire totale ont rarement été rapportés [15]. Par ailleurs, un infiltrat inflammatoire dans la lamina propria a été observé dans de nombreux cas. Cet infiltrat est composé essentiellement de plasmocytes et d’éosinophiles [10, 14, 15], et dans certains cas d’un grand nombre de mastocytes [10].
Étant donné les résultats des études radiologiques au cours de la MS, les lésions intestinales macroscopiques sont probablement fréquentes chez les patients. Dans une étude par transit œsogastro-duodénal et transit du grêle ayant porté sur 78 patients atteints de MS [16], l’anomalie la plus fréquemment observée (73 % des patients) était la présence de nodules de 1 à 3 mm dans la muqueuse de l’intestin grêle, correspondant probablement à de l’œdème. Dans une seconde étude, portant sur 14 patients [9], il était observé dans 57 % des cas un épaississement de la muqueuse intestinale, des lésions nodulaires ou polypoïdes.
En dehors de la diarrhée, des symptômes liés à une atteinte colique de la MS ne sont pas rapportés. De plus, l’implication d’une atteinte colique dans la pathogénie de la diarrhée au cours de la MS n’est pas claire. Les lésions coliques observées sont variables : nodules muqueux de petites tailles [16], œdème muqueux éventuellement accompagné de lésions urticariennes, lésions polypoïdes [17]. Histologiquement, ces lésions comportent souvent un infiltrat inflammatoire qui peut être composé de polynucléaires éosinophiles et de mastocytes, ou uniquement de mastocytes. Dans certains cas, des ulcérations coliques ont été rapportés, faisant porter le diagnostic erroné de maladie de Crohn [17]. Il faut noter qu’il n’a pas été évalué si la présence de lésions coliques était corrélée à l’existence d’une diarrhée au cours de la MS. Les symptômes gastro-intestinaux sont aussi fréquents que le prurit chez les patients atteints de mastocytose systémique
Pathogénie des symptômes gastro-intestinaux au cours de la mastocytose systémique
La pathogénie de la symptomatologie gastro-intestinale au cours de la MS n’est pas claire dans la majorité des cas. La rareté et le caractère polymorphe de cette maladie rendent les études prospectives sur un grand nombre de patients difficiles.
Les douleurs abdominales ont parfois été attribuées à une maladie ulcéreuse, à une infiltration mastocytaire du tube digestif ou à la libération de médiateurs mastocytaires [5]. Les douleurs abdominales peuvent être de types dyspeptiques ou non dyspeptiques et les deux types de douleurs peuvent être présents chez le même patient [7, 9]. Dans l’étude prospective de Cherner [7], les patients ayant des douleurs abdominales étaient soulagés par les anti-acides (anti-H2) dans 56 % des cas, ce qui suggère que l’hypersécrétion acide gastrique a un rôle important dans la pathogénie de la douleur chez ces patients. Cette hypersécrétion gastrique est très probablement liée à l’hyperhistaminémie, comme le montre notamment la corrélation entre le taux d’histamine et la sécrétion acide dans l’étude de Cherner [7]. En revanche, il n’existe aucun argument pour un rôle d’une éventuelle hypergastrinémie [5].
La pathogénie de la diarrhée est mal comprise et possiblement différente d’un patient [11] à l’autre. Chez certains patients, l’hypersécrétion gastrique est un élément majeur contribuant à la diarrhée [7]. Chez ces patients, le mécanisme par lequel l’hypersécrétion gastrique cause la diarrhée est probablement similaire à celui du syndrome de Zollinger-Ellison dans lequel l’hyperacidité inactive les enzymes pancréatiques et affecte directement les villosités intestinales.
Chez d’autres patients, une surproduction de prostaglandine D2 (PGD2) pourrait être un facteur important dans la pathogenèse de la diarrhée. La production de PGD2 peut être transitoirement augmentée chez certains patients atteints de MS [5] et, dans certains cas, un traitement par aspirine, qui inhibe la synthèse de prostaglandine, a une grande efficacité sur la diarrhée [18]. Néanmoins, l’aspirine pouvant induire la dégranulation des mastocytes, il est préférable dans cette indication de débuter l’aspirine à faible dose et d’y associer des antagonistes des récepteurs H1 et H2 et probablement un traitement par inhibiteur de la pompe à proton. La fréquence du rôle d’une surproduction de PGD2 dans la diarrhée au cours de la MS n’est pas établie.
L’infiltration de l’intestin grêle ou du côlon par des mastocytes pourrait également jouer un rôle chez certains patients. Cependant, les rôles respectifs de l’infiltration mastocytaire par elle-même, des secrétions mastocytaires, des altérations architecturales induites (atrophie villositaire partielle, lésions urticarienne…) ou de l’association de ces facteurs n’est pas claire, d’autant plus qu’aucune corrélation entre diarrhée et lésion histologique de l’intestin grêle et du côlon n’a été réalisée à ce jour.
Enfin, le rôle d’une accélération du transit induite par la libération de médiateurs mastocytaires a été évoqué [5] mais reste à confirmer.
La malabsorption n’est pas fréquente au cours de la MS et elle est le plus souvent minime, bien que certains auteurs aient rapporté de rares cas avec des formes sévères [15]. Chez ces patients, la fonction pancréatique est normale à chaque fois qu’elle est explorée, et la malabsorption est liée à une atteinte intestinale, avec fréquemment une perturbation des tests fonctionnels et des anomalies histologiques (villosités émoussées, infiltrat inflammatoire composé ou non de mastocytes en excès) [5]. L’hypersécrétion acide gastrique joue possiblement un rôle dans certains cas de malabsorption car une normalisation de l’absorption intestinale a été observée après correction de l’acidité gastrique chez certains patients. En revanche, certains cas de malabsorption ne sont clairement pas liés à l’hyperacidité gastrique, celle-ci étant absente ou les symptômes persistant malgré la normalisation de la sécrétion acide par l’utilisation d’anti-sécrétoires. Enfin, de rares cas d’entéropathie sensible au gluten ont été rapportés au cours de la MS [14, 15], mais ce mécanisme n’est pas impliqué chez la grande majorité des patients présentant une malabsorption. La pathogénie de la symptomatologie gastro-intestinale au cours de la mastocytose systémique est attribuée à une infiltration mastocytaire du tube digestif ou à la libération de médiateurs mastocytaires
Diagnostic d’une atteinte gastro-intestinale au cours d’une mastocytose systémique
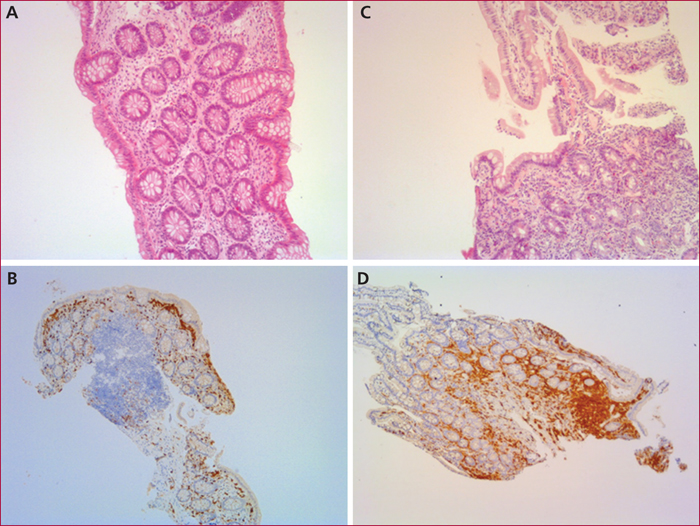
Chez un patient ayant une MS déjà connue, la fréquence des symptômes digestifs est très élevée comme nous l’avons vu plus haut. Lors de l’exploration de ces symptômes, des biopsies devront être réalisées au cours des endoscopies afin de chercher une infiltration mastocytaire. La détection des mastocytes devra se faire à l’aide des techniques d’immunohistochimie car ils sont difficilement détectables avec les colorations standard (figure 1). Les mastocytes expriment le CD117 (c-Kit) et la tryptase, et lorsqu’ils sont anormaux le CD25 et le CD2. L’absence d’infiltration mastocytaire gastro-intestinale n’élimine pas l’origine mastocytaire des symptômes car, comme nous l’avons vu, la libération de médiateurs par des mastocytes à distance peut induire des symptômes digestifs.
La symptomatologie gastro-intestinale n’est pas spécifique et peut souvent avoir un retentissement important sur la qualité de vie des patients sans pour autant donner lieu à des signes objectifs de dysfonction gastro-intestinale. En l’absence d’autres signes systémiques et en particulier cutanés (les signes cutanés sont absents jusqu’à 50 % des cas dans certaines séries de MS [8]), de tels symptômes peuvent être compatibles avec le diagnostic de trouble fonctionnel gastro-intestinal. Les patients ayant une symptomatologie invalidante mais compatible avec un trouble fonctionnel gastro-intestinal ont souvent des explorations biologiques, radiologiques et endoscopiques. Bien qu’aucune recommandation officielle n’existe, il pourrait être intéressant chez ce type de patients de réaliser un dosage de la tryptase sérique (en même temps que le bilan biologique de débrouillage souvent réalisé) et d’encourager les anatomopathologistes à chercher les mastocytes sur les biopsies gastro-intestinales réalisées à l’aide d’un marquage CD117/C-KIT. En fonction du résultat de ces examens, on pourra discuter de l’intérêt d’une biopsie ostéo-médullaire et de la recherche de la mutation D816V de C-KIT afin d’affirmer le diagnostic de MS.
Certaines maladies gastro-intestinales malignes peuvent être confondues avec une MS. Il faut en effet savoir évoquer le diagnostic de MS devant une forme atypique de syndrome de Zollinger-Ellison, de syndrome carcinoïde ou de VIPome. Ces trois syndromes peuvent causer des douleurs abdominales, une diarrhée et une malabsorption. Par ailleurs, comme au cours de la MS, des flushes peuvent être observés au cours d’un syndrome carcinoïde ou d’un VIPome, et des ulcères gastro-duodénaux au cours du syndrome de Zollinger-Ellison. Certaines particularités cliniques permettent de distinguer la MS de ces trois autres syndromes : les flushs de la MS ne s’accompagnent pas de sueurs ; une urticaire pigmentaire est fréquente au cours de la MS alors qu’un rash est rarement observé au cours des trois autres syndromes ; la diarrhée du VIPome est beaucoup plus abondante que celle observée en cas de MS (souvent supérieure à 2 L/24 h contre inférieure à 1 L/24 h) ; une hépato-splenomégalie est observée dans près de 20 % des MS alors qu’elle est beaucoup plus rare dans les trois autres cas. Par ailleurs, les dosages biologiques permettent de confirmer ou d’infirmer le diagnostic : la gastrinémie, le taux sérique de VIP et la sérotoninémie (ainsi que les 5HIAA urinaires) sont élevés en cas de syndrome de Zollinger-Ellison, de VIPome et de syndrome carcinoïde respectivement, alors que ces dosages sont normaux dans la MS. Il faut savoir évoquer le diagnostic de mastocytose systémique devant une forme atypique de syndrome de Zollinger-Ellison, de syndrome carcinoïde ou de VIPome
Traitement de la symptomatologie gastro-intestinale au cours de la mastocytose systémique
La MS nécessite une adaptation thérapeutique à chaque profil de patient. Compte tenu de sa faible prévalence, le traitement est essentiellement fondé sur l’expérience et comporte deux grands axes. Le premier vise à prévenir et à limiter la dégranulation et/ou ses conséquences ; c’était, jusqu’à très récemment, le seul traitement que l’on pouvait proposer. Le second a pour objectif de contrôler la prolifération tumorale mastocytaire. Les récents progrès accomplis dans la compréhension de la physiopathologie ont permis de nouvelles approches thérapeutiques, notamment avec l’interféron alpha et la cladribine. Plus récemment, des inhibiteurs de tyrosine kinase ont vu le jour et d’autres sont en cours de développement afin d’inhiber l’activité tyrosine kinase de C-KIT et ainsi bloquer l’activation, la migration et la survie des mastocytes pathologiques. L’indication et la mise en place de ce type de traitement doivent être réservées aux hématologues. Les mesures générales et les traitements symptomatiques restent classiques, utilisant les antihistaminiques, le cromoglycate disodique, plus rarement l’aspirine, les antagonistes des récepteurs des leucotriènes [1]. On peut, lors d’importantes crises douloureuses abdominales, utiliser le budésonide (Entocort®, dose maximale 9 mg/j) qui a un faible effet systémique, ce qui limite les effets indésirables. La corticothérapie générale peut aider au contrôle d’une diarrhée avec malabsorption ou de crises douloureuses abdominales par poussées œdémateuses. Instituée à la dose initiale de 1 mg/kg, sa décroissance doit être rapide, son utilisation chronique pouvant aggraver une ostéoporose fréquente dans la mastocytose. Les traitements cytoréducteurs sont indiqués en cas de maladie agressive avec dysfonction d’organe liée à l’infiltration mastocytaire. L’interféron peut diminuer les symptômes digestifs de façon significative mais est souvent mal toléré, notamment d’un point de vue psychiatrique. Les chimiothérapies classiques même à fortes doses sont inefficaces. La cladribine (2CdA, Leustatine®), un analogue nucléosidique, permet d’améliorer les symptômes digestifs dans plus de 60 % des cas (données non publiées). Cependant les réponses sont souvent transitoires et des cures successives sont souvent nécessaires. Leur nombre et leur fréquence doivent être mis en balance avec le risque myélotoxique et immunosuppresseur du produit. Les espoirs récents portent sur les inhibiteurs de tyrosine kinase avec des effets thérapeutiques à la fois sur les symptômes et l’infiltration mastocytaire. Le traitement comporte deux grands axes : prévenir et limiter la dégranulation et/ou ses conséquences, contrôler la prolifération tumorale mastocytaire
Conclusion
Les mastocytoses systémiques constituent un ensemble d’affections hétérogènes autant par leur présentation clinique et biologique que par leur pronostic ou leur traitement. La forte prévalence des manifestations gastro-intestinales et surtout leur retentissement fonctionnel important méritent de les caractériser et de les prendre en charge afin d’améliorer la qualité de vie des patients. D’autre part, le diagnostic de MS avec atteinte gastro-intestinale devrait être évoqué devant un tableau de trouble fonctionnel intestinal sévère. Des examens simples (tryptase sérique et immuno-histochimie sur les biopsies gastro-intestinales), complétant le bilan biologique et morphologique réalisé habituellement dans cette situation, pourraient permettre de redresser le diagnostic.Take home messages
Liens d’intérêts
l’auteur déclare n’avoir aucun lien d’intérêt en rapport avec l’article.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License