Hématologie
MENU19 Hémopathies | divers Volume 26, supplement 4, Septembre 2020

19-01
Implication de la voie de signalisation Hedgehog dans la mastocytose
L. Polivka*1, E. Bayard2, M. Tissandier3, P. Véronique4, E. Soucié5, B. Bader-Meunier6, S. Fraitag7, D. Canioni7, L. Lhermitte8, J. Rossignol9, M. Arock10, P. Dubreuil11, C. Bodemer12, O. Hermine13, L. Maouche-Chrétien14
1 Hôpital Necker AP-HP, Paris ; 2 U1163- hématologie, Institut Imagine, Paris ; 3 U1163 - hématologie, Institut Imagine, Paris ; 4 Animalerie, Hôpital Saint-Louis AP-HP, Paris ; 5 Hématologie, CRCM, Marseille ; 6 Unité d’immuno-hémato et rhumatologie pédiatrique, Hôpital Necker, Paris ; 7 Laboratoire de pathologie, Hôpital Necker, Paris ; 8 Laboratoire hématologie biologique, Hôpital Necker, Paris ; 9 Hématologie adulte, Hôpital Necker, Rue de Sèvres, Paris, France, Paris ; 10 Pharmacie, École normale supérieure de Cachan, Paris ; 11 Crcm, U1068 Inserm, Marseille ; 12 Dermatologie, Hôpital Necker, Paris ; 13 Hématologie/centre de référence de l’hémophilie, AP-HP, Paris ; 14 Hématologie, Institut Imagine, Paris
Introduction. La mastocytose est une affection néoplasique caractérisée par l’accumulation de mastocytes dans un ou plusieurs organes. Des mutations somatiques activatrices du gène KIT ont été identifiées chez 85 % des patients. La mutation D816V est la plus fréquemment retrouvée et est associée à une activation constitutive du récepteur à activité tyrosine kinase KIT dans les mastocytes pathologiques. Mais cette mutation n’explique pas à elle seule l’expansion clonale des mastocytes et l’hétérogénéité des présentations cliniques observées. Sur 2400 patients enregistrés au centre de référence des mastocytoses (CEREMAST), 8 % présentent une forme familiale et une minorité d’entre eux ont une mutation germinale dans le gène KIT (autre que D816V, létale in utero). Toutes les tentatives visant à démontrer l’effet oncogène de la mutation KITD816V ont échoué, et moins de 50 % des formes agressives répondent aux inhibiteurs de l’activité tyrosine kinase de KITD816V, avec un succès limité. Il est donc clair que d’autres facteurs coopèrent avec la voie KIT pour le développement de la pathologie. L’objectif de notre étude était d’identifier dans des formes rares de mastocytoses (mastocytoses agressives congénitales ou formes familiales) des événements génétiques primitifs agissant en synergie avec la mutation de KIT, pour en éclaircir les mécanismes physiopathologiques et les tester dans les formes sporadiques.
Résultats. Nous avons identifié chez trois patients issus de deux familles indépendantes qui présentaient l’association : i)- d’une mastocytose congénitale agressive (patient #1) ou familiale (patient #2 et #3) et ii)- d’un syndrome de céphalopolysyndactylie de Greig – syndrome rare associant une macrocéphalie et une polydactylie – des mutations hétérozygotes perte de fonction dans le gène GLI3, codant pour le principal facteur de transcription répresseur de la voie Hedgehog (HH) : GLI3R. La coexistence de ces deux événements rares, nous a menés à étudier le rôle de la voie HH dans la physiologie du mastocyte et dans la physiopathologie de la mastocytose, et notamment des formes sporadiques agressives. Nous avons démontré pour la première fois que la voie HH était activée dans les mastocytes primaires normaux et constitutivement hyperactivée dans les mastocytes néoplasiques de patients atteints de mastocytoses agressives et dans toutes les lignées de mastocytes mutées pour KIT. Nous avons montré que l’utilisation d’inhibiteurs de la voie de signalisation HH inhibe la prolifération et induit l’apoptose des mastocytes néoplasiques primaires issus de patients, et retarde significativement la mort chez les souris présentant une mastocytose agressive KITD816V+. Pour confirmer la capacité de l’haplo-insuffisance de GLI3 à induire les tumeurs, nous avons utilisé des souris GLI3Xt+/-, modèle murin du syndrome de Greig, et nous avons montré que les BMMC dérivés de ces souris et transduits avec le vecteur KITD816V avaient un fort potentiel prolifératif par rapport aux témoins, confirmant la coopération oncogénique entre les voies HH et KIT. Nous avons également montré que l’expression ectopique de GLI3R induisait la mort des mastocytes en quelques jours, confirmant son rôle de gène suppresseur de tumeur dans le mastocyte.
Conclusion. Ainsi, à partir de phénotypes extrêmes de mastocytoses dans des contextes syndromiques germinaux particuliers, nous avons pu mettre en évidence un nouveau mécanisme impliqué dans la physiopathologie des mastocytoses agressives en général et identifier une nouvelle cible thérapeutique pour cette pathologie.
19-02
Cytologie 2.0 : développement et validation d’un algorithme d’intelligence artificielle pour l’étude cytologique des hyperlymphocytoses
M. Sahasrabudhe1, M. Vakalopoulou1, E. Zacharaki2, B. Grange3, L. Jallades3, N. Paragios1, P. Sujobert*3
1 Centre de vision numérique, CentraleSupélec - Université Paris-Saclay, Gif-sur-Yvette ; 2 Division of Telecommunications and Information Technology, Université de Patras, Patras, Grèce ; 3 Hématologie, CH Lyon Sud, Pierre-Bénite
Introduction. La cytologie est une discipline complexe, dont l’apprentissage est long et difficile, et qui est caractérisée par une assez grande variabilité interopérateur. Les techniques d’intelligence artificielle basées sur le deep learning arrivent à des performances proches des experts pour l’analyse d’images médicales. Nous rapportons ici la mise au point et le test d’un outil permettant l’analyse cytologique des hyperlymphocytoses.
Matériels et méthodes. Mise au point de l’outil : 209 cas d’hyperlymphocytose supérieure à 4 G/L ont été inclus, en recueillant à chaque fois les données démographiques, la valeur de la lymphocytose, les images de lymphocytes acquises de manière standardisée par un DM96. Pour chaque patient, un immunophénotypage a été réalisé pour affirmer de manière certaine la nature tumorale ou réactionnelle de l’échantillon. Ces 209 cas ont été séparés en une cohorte d’entraînement (n = 146), une cohorte de validation (n = 21) et une cohorte de test (n = 42). Plusieurs techniques de deep learning ont été évaluées, pour résoudre notamment une difficulté particulière liée au fait que l’annotation portait sur l’ensemble du frottis sanguin (normal ou pathologique) mais pas sur chaque cellule prise individuellement. Ces différents algorithmes ont été comparés aux performances de 12 biologistes interprétant les mêmes données (images issues du DM96). De plus, des tests mesurant la répétabilité et la reproductibilité de l’outil ont été effectués, qui seront communiqués lors de la conférence.
Test de l’outil : 39 nouveaux cas ont été soumis à l’algorithme dans sa version finalisée, pour attribuer un score continu allant de 0 (réactionnel) à un (tumoral). Les frottis sanguins de ces patients ont été ensuite présentés à un panel de 10 cytologistes expérimentés, en randomisant pour chaque cas et chaque biologiste si le biologiste avait accès ou non au résultat de l’algorithme. Il était demandé aux biologistes de classer les cas en réactionnel ou tumoral. La performance globale des biologistes sans ou avec l’aide de l’algorithme était l’objectif principal de cette étude.
Résultats. En utilisant une technique dite de multiple instance learning, nous avons mis au point un algorithme dont les performances sont meilleures que celles des biologistes (précision 88 % vs 77 %) sur les images de lymphocytes numérisées par le dispositif DM96.
Dans la phase de test de 39 cas (14 tumoraux et 25 réactionnels), l’utilisation de l’algorithme permettait d’améliorer les performances des biologistes pour classer les patients d’après l’analyse du frottis sanguin (84 % de réponses correctes vs 80,5 % sans l’algorithme). De manière intéressante, les cinq biologistes les plus expérimentés voyaient leurs performances diminuées par l’utilisation de l’algorithme (78 % vs 81 %) alors que l’on observait au contraire un effet bénéfique marqué pour les cinq biologistes les moins expérimentés (90 % vs 80 %).
Conclusion. Cette étude montre qu’il est possible d’améliorer le diagnostic cytologique des hyperlymphocytoses en utilisant des outils d’intelligence artificielle. Cependant, il est indispensable de préciser dans quelles conditions ces outils doivent être utilisés, car nous avons observé des différences entre les biologistes selon leur niveau d’expérience. L’intérêt de ces approches pour des problèmes cytologiques plus complexes devra faire l’objet de nouvelles études.
19-03
Cytologie médullaire des thrombopénies familiales avec prédisposition aux leucémies aiguës et syndromes myélodysplasiques
E. Fournier1, V. Soenen1, N. Trillot2, F. Gonzales3, V. Tintiller2, L. Terriou4, C. Derrieux1, W. Abou Chahla3, C. Paris2, C. Berthon5, T. Boyer6, A. Lambliotte3, S. Susen7, C. Preudhomme1, N. Duployez*1
1 Laboratoire d’hématologie, CHU Lille, Lille ; 2 Hématologie - transfusion, CHU Lille, Lille ; 3 Hématologie pédiatrique, CHU Lille, Lille ; 4 Unité d’immunologie clinique, EA2686, CHU Lille, Lille ; 5 Maladies du sang, CHU Lille, Lille ; 6 Laboratoire d’hématologie, CHU Amiens-Picardie, Lille ; 7 Pole hématologie-transfusion, CHU Lille, Inserm Laboratoire J&K, U1011, U995, Lille
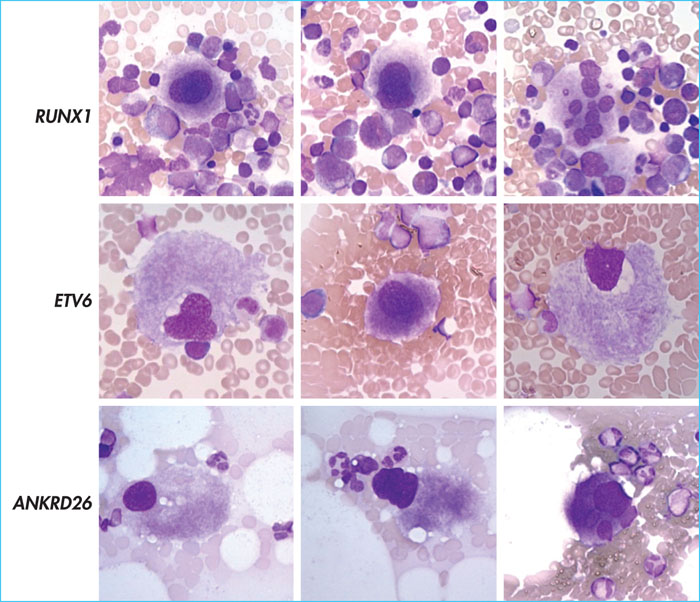
Introduction. Les mutations germinales des gènes RUNX1, ETV6 et ANKRD26 sont responsables de certaines formes de thrombopénies congénitales à transmission autosomique dominante avec prédisposition au développement d’hémopathies malignes (leucémies aiguës et syndromes myélodysplasiques essentiellement). Les cancers hématologiques associés à ces anomalies génétiques sont maintenant reconnus comme une entité à part entière dans la classification OMS 2016 des néoplasies myéloïdes et des leucémies aiguës.
Peu d’études se sont intéressées aux aspects morphologiques de la moelle osseuse chez les patients présentant une mutation constitutionnelle sous-jacente en dehors du contexte d’hémopathie.
Patients et méthodes. Nous rapportons ici la cytologie médullaire chez cinq individus diagnostiqués au CHU de Lille présentant une thrombopénie avec prédisposition génétique aux hémopathies (RUNX1, n = 3; ETV6, n = 1 et ANKRD26, n = 1) en l’absence de cancer hématologique actif.
Résultats. La moelle osseuse des cinq individus présentait une dysmégacaryopoïèse caractéristique avec de petits mégacaryocytes à noyaux hypo- ou unilobés sans atteinte des autres lignées (figure). La richesse en mégacaryocytes était normale. Les trois individus avec mutation germinale de RUNX1 présentaient également quelques mégacaryocytes avec des noyaux à lobes séparés (figure 1).

Conclusion. Ces observations soulignent l’importance de l’examen initial de la moelle osseuse afin d’établir les caractéristiques cytologiques qui serviront de référence pour le suivi. Elles démontrent la nécessité de critères spécifiques pour le diagnostic de syndrome myélodysplasique chez les individus présentant une thrombopénie congénitale avec prédisposition aux hémopathies malignes.
19-04
Mutations germinales de TET2 et prédisposition aux hémopathies myéloïdes : à propos d’une famille
N. Duployez*1, L. Goursaud2, L. Fenwarth1, C. Bories3, A. Marceau-Renaut1, T. Boyer4, E. Fournier1, O. Nibourel1, C. Roche-Lestienne5, G. Huet6, D. Beauvais2, C. Berthon2, N. Cambier7, B. Quesnel2, C. Preudhomme1
1 Laboratoire d’hématologie, CHU Lille, Lille ; 2 Maladies du sang, CHU Lille, Lille ; 3 Hématologie, CH Lens, Lille ; 4 Laboratoire d’hématologie, CHU Amiens-Picardie, Lille ; 5 Laboratoire de cytogénétique, CHU Lille, Lille ; 6 Culture cellulaire, CHU Lille, Lille ; 7 Service d’hématologie clinique, CH De Valenciennes, Valenciennes
Introduction. Le gène TET2 code une méthylcytosine dioxygénase catalysant la conversion de la 5-méthylcytosine en 5-hydroxy-methylcytosine permettant la déméthylation de l’ADN et la modulation de l’expression génique. Le gène TET2 est l’un des gènes les plus fréquemment mutés dans les hémopathies myéloïdes (40-50 % des leucémies myélomonocytaires chroniques [LMMC], 20-25 % des syndromes myélodysplasiques [SMD] et leucémies aiguës myéloïdes [LAM] de novo, 10-15 % des syndromes myéloprolifératifs [SMP] Ph-négatifs). L’étude de modèles murins tet2-KO a permis de démontrer que son haplo-insuffisance conférait une susceptibilité au développement de pathologies myéloïdes.
Patients et méthodes. Nous rapportons le cas de trois frères et sœurs ayant développé LAM, LMMC et polyglobulie de Vaquez (PV) à l’âge de 61, 60 et 53 ans respectivement. Les trois individus présentaient tous des antécédents de troubles thyroïdiens ayant conduit à une thyroïdectomie partielle ou totale. Leur mère était décédée d’un lymphome de type T diagnostiqué à 67 ans.
Résultats. Le séquençage ciblé d’un panel de 36 gènes chez les trois individus a montré qu’ils partageaient un variant frameshift de TET2 (K1363fs). Le variant était également retrouvé sur culture de fibroblastes de l’une des filles. Cette mutation est supposée conduire à la production d’une protéine tronquée avec disparition du deuxième domaine DSBH (double-stranded β-helix) de la protéine TET2, critique pour la liaison de la protéine à son substrat et à la 5-méthylcytosine. Une protéine TET2 sans ce domaine est prédite comme instable et rapidement dégradée conduisant à une haplo-insuffisance.
Discussion. Les mutations germinales de TET2 ont été récemment décrites dans une famille présentant de multiples cas de lymphomes (Kaasinen et al, Nat Commun, 2019). Notre observation suggère que les mutations germinales de TET2 prédisposent au développement d’hémopathies myéloïdes. Le phénotype variable des pathologies hématologiques observées est probablement en rapport avec les anomalies génétiques acquises au moment de la transformation (SRSF2, JAK2…) et/ou les autres facteurs génétiques constitutionnels des individus. Récemment, de larges études de séquençage ont montré que TET2 était une cible mutationnelle dans certains cancers thyroïdiens. De nouvelles études seront nécessaires pour confirmer ou infirmer si les mutations germinales de TET2 sont liées aux troubles thyroïdiens observés dans cette famille (une autre anomalie germinale ne pouvant être exclue).
Conclusion. Il est admis que la fréquence des prédispositions aux hémopathies malignes est largement sous-estimée. Nous décrivons ici un nouveau syndrome de prédisposition aux hémopathies myéloïdes caractérisé par un âge de transformation similaire à celui observé dans les cas sporadiques. L’incidence des mutations germinales de TET2 reste difficile à déterminer en l’absence de prélèvement constitutionnel systématique puisque les mutations somatiques de TET2 restent généralement positives chez les patients en rémission de leur maladie, compatible avec un état préleucémique.
19-05
Colites à l’idélalisib : étude rétrospective monocentrique en vie réelle et analyse des données de la pharmacovigilance française
C. Breal*1, T. Witasse2, F. Beuvon3, S. Dermine4, P. Franchi-Rezgui1, B. Deau-Fisher1, L. Willems1, E. Grignano1, A. Contejean1, D. Bouscary1, JM. Treluyer2, JL. Faillie5, C. Guerin6, L. Chouchana2, M. Vignon1
1 Hématologie, Assistance Publique Hôpitaux de Paris, Paris ; 2 Pharmacovigilance, Hôpital Cochin, Paris ; 3 Anatomopathologie, Hôpital Cochin, Paris ; 4 Gastro-entérologie, Hôpital Cochin, Paris ; 5 Pharmacovigilance, Hôpital Lapeyronie, Montpellier ; 6 Pharmacie, Hôpital Cochin, Paris
Introduction. L’idélalisib (I) est un médicament oral inhibiteur des phosphatidylinositol 3-kinase delta indiqué dans le traitement de la leucémie lymphoïde chronique (LLC) et des lymphomes B non hodgkiniens (LNH-B) en rechute. La colite secondaire à l’idélalisib (CI) est un de ses principaux effets secondaires, estimée à 15 % dans la littérature.
Patients et méthodes. L’ensemble des dossiers des patients avec dispensation hospitalière initiale d’I au sein d’un centre ont été rétrospectivement revus, ainsi que tous les cas de CI rapportés au Réseau français de pharmacovigilance (RFPV). La période d’étude est entre 2014 et 2018.
Résultats. Dans notre centre, 15 patients ont été traités par I (neuf hommes et six femmes) : huit (53 %) pour un LNH-B et sept (47 %) pour une LLC. L’âge médian à l’introduction d’I était de 76 ans (55-89). Le nombre de lignes antérieures de traitement était de 3,5 (1-7). L’I était administré seul (20 % des cas) ou associé au rituximab (R) (47 %) ou à l’ofatumumab (Ofa) (27 %). Six patients (40 %) ont développé une CI dans un délai médian de quatre mois (3-6). La sévérité était de grade ≥ 3 et trois patients ont nécessité une hospitalisation en réanimation. Toutes les CI ont entraîné un arrêt de l’I. Deux patients ont évolué favorablement sous traitement symptomatique et quatre (67 %) ont nécessité une corticothérapie. Les coprocultures avec recherche de Clostridium difficile étaient négatives. Une endoscopie a été réalisée dans tous les cas. La macroscopie était anormale dans 2/3 des cas avec des signes d’inflammation à l’histologie dans cinq cas. Le scanner était anormal pour trois des quatre réalisés. Le traitement par I a été repris après résolution de la diarrhée pour trois patients avec réapparition de la CI chez deux patients, dont un cas conduisant au décès. Deux patients sont décédés de leur pathologie hématologique, en absence d’alternative. Les caractéristiques des patients étaient comparables dans les groupes CI et absence de CI, sauf le nombre de lignes de traitement antérieur (4,5 vs 2,9) ainsi que l’association à l’Ofa (50 % vs 11 %) plus important dans le groupe CI.
Parmi les cas notifiés au RFPV, 50 cas sont compatibles avec un tableau de colite sous idélalisib, dont quatre avec une documentation bactériologique positive. Un total de 46 cas de CI (32 hommes et 14 femmes) a été inclus, dont 43 (94 %) de grade 3-4. L’âge médian était de 71 ans (52-89). L’idélalisib était prescrit pour une LLC chez 26 patients (59 %), pour un LNH-B chez 20 patients (43 %), en monothérapie pour 27 patients (57 %) et en association au R et à l’Ofa pour 16 (33 %) et trois (7 %) patients. Il n’y avait pas d’autre médicament susceptible d’induire une colite chez 38 (83 %) patients. Le délai médian d’apparition des signes cliniques était de 122 jours (15-427). 19 patients ont eu une coloscopie avec biopsies. La macroscopie était anormale chez 16 (84 %) patients et l’histologie était en faveur d’un aspect de colite inflammatoire aspécifique. Le délai médian de prise en charge spécifique de la CI était de 20 jours (1-548). Le traitement a été stoppé chez 44 patients (96 %) et 24 patients (49 %) ont été traités par corticothérapie.
Conclusion. En situation de vie réelle, 40 % des patients traités par idélalisib ont eu une CI. Cette complication est tardive (délai médian de quatre mois) et sévère (grade ≥ 3). Elle entraîne le plus souvent un arrêt du traitement. Ces données concordent avec celles de la pharmacovigilance. L’identification précoce de cet effet indésirable grave peut améliorer sa prise en charge et le pronostic hématologique des patients.
19-06
Le syndrome d’activation macrophagique : étude rétrospective de 110 cas
R. Hassine*1, R. Mezrigui1, L. Khefacha2, S. Boukhris3, R. R.3, S. Chouchene1, R. Marwa1, M. Sassi2, W. Guetari1, A. Laatiri3, M. Hassine1
1 Laboratoire d’hématologie et banque du sang, CHU Fattouma Bourguiba Monastir, Monastir, Tunisie ; 2 Laboratoire de biologie, EPS Fattouma Bourguiba, Centre de Maternité et de Néonatologie, Monastir, Tunisie ; 3 Hématologie clinique, CHU Fattouma Bourguiba, Monastir, Tunisie
Introduction. Le syndrome d’activation macrophagique (SAM), ou syndrome d’activation lymphohistiocytaire, est une maladie rare mais potentiellement fatale traduisant un désordre du système immunitaire. C’est la conséquence d’une stimulation inappropriée de lymphocytes cytotoxique et de macrophages, entraînant une phagocytose anormale des cellules hématopoïétiques. Le diagnostic reste difficile et repose sur un faisceau d’arguments cliniques et biologiques. Le but de notre travail est de décrire les aspects clinicobiologiques du SAM chez une population tunisienne.
Matériels et méthodes. Il s’agit d’une étude rétrospective sur la période de cinq ans (2015-2019). Les patients présentant des images d’hémophagocytose au niveau du myélogramme ont été retenus. La fiche de renseignement accompagnant les demandes de myélogramme a servi de source de données, lesquelles ont été traitées par le logiciel SPSS Statistics 20.
Le diagnostic du SAM était retenu sur les critères du groupe français de lymphohistiocytose hémophagocytaire par l’association de cinq critères parmi huit, dont deux ne sont pas réalisés en Tunisie.
Résultats. 110 cas ont été colligé : 58 hommes et 52 femmes. L’âge médian est de 34,17 ans (min = 3 mois, max = 81 ans). La majorité des prélèvements provenaient des services de l’hôpital de Mahdia et du service de Pédiatrie, avec respectivement 25,5 % et 15,5 % des prélèvements.
Les données complètes de l’hémogramme ont été fournies pour 90 patients, dont les deux tiers présentaient une bi- ou une pancytopénie. La fièvre a été présente dans 76,3 % des cas et la splénomégalie dans 27 % des cas. Quand les bilans biochimiques ont été rapportés sur la fiche de renseignements, on notait une hyperferritinémie dans 36 cas, une hypertriglycéridémie dans 21 cas, et une hypofibrinogénémie dans sept cas seulement.
Seulement 15 patients répondaient aux critères de diagnostic avec un nombre de critères ≥ cinq. Il était de quatre pour 21,8 % des patients et de trois pour 28,2 % des patients.
Discussion. Mis à part le manque des données cliniques et biologiques, on constate que la présence d’image d’hémophagocytose peut orienter vers le diagnostic d’un SAM en absence des autres éléments du diagnostic. Devant la mortalité importante de ce syndrome en cas de diagnostic tardif, le clinicien n’attend généralement pas la réunion de cinq critères pour poser le diagnostic et démarrer le traitement.
Conclusion. Le SAM reste une entité clinicobiologique difficile à diagnostiquer. Un bon jugement clinique et une interprétation minutieuse de tous les critères biologiques et cliniques continuent à être la base du diagnostic, notamment la recherche d’images d’hémophagocytose dont la présence signe presque toujours le diagnostic en pratique clinique courante.
19-07
La mesure de potentiel de membrane : une méthode simple et fiable pour la caractérisation des canalopathies
S. Egée*1, AD. Monedero1, L. Pérès1, E. Fermo2, L. Kaestner3, R. Van Wijk4, P. Bianchi5
1 Équipe physiologie comparée des érythrocytes, UMR8227 -CNRS-Université Pierre et Marie Curie, Roscoff ; 2 Uoc Ematologia, uos Fisiopatologia delle Anemie, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italie ; 3 Theoretical Medicine and Biosciences, Université de la Sarre, Sarrebruck, Allemagne ; 4 Department of Clinical Chemistry and Hematology, UMC Utrecht, Utrecht, Pays-Bas ; 5 Uoc Ematologia, uos Fisiopatologia delle Anemie, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico - Clinica Regina Elena, Milano, Italie
Introduction. L’état d’hydratation des érythrocytes est important pour maintenir leurs propriétés rhéologiques dans la circulation sanguine et assurer ainsi l’efficacité du transport des gaz. Le contenu en eau des globules rouges est dicté par les concentrations en cations intracellulaires dont l’équilibre est maintenu par l’équilibre entre les activités de fuite et de pompes. Néanmoins, une série d’études récentes ont montré que plusieurs troubles érythrocytaires héréditaires de l’homéostasie de volume se produisent lorsque des canaux ioniques tels que le canal Gárdos ou le canal mécanosensible PIEZO1 sont mutés.
L’objectif de notre travail a été de rechercher et d’établir une méthode simple afin de permettre la caractérisation fonctionnelle des différentes mutations pour ces canaux sans avoir recours à l’expression hétérologue.
Résultats. Dans le cadre de cette étude, la méthode CCCP a été validée sur des cellules de patients pour lesquelles des informations fonctionnelles étaient déjà disponibles (Gardos Hst R352H, Piezo1 HX R2456H) démontrant ainsi la faisabilité de l’utilisation de cette technique pour caractériser les différents variants. La mutation R352H sur le canal Gárdos se situe dans la région du site de fixation de la calmoduline (CaMDB2) laissant présager une modification de la sensibilité du canal pour le calcium intracellulaire. Un agoniste du canal Gárdos, le NS309, a pour effet de diminuer la concentration de calcium requise pour éliciter son activité. En l’utilisant, nous avons pu démontrer que la mutation décrite augmente d’au moins un ordre de grandeur la sensibilité du canal muté aux faibles concentrations de calcium intracellulaire.
Soumettre les globules rouges à une solution de faible force ionique (LIS) est connu pour provoquer une dépolarisation instantanée, atteignant des valeurs de potentiel de membrane très positives. Dans ces conditions, un canal cationique non sélectif et un canal de chlorure s’ouvrent pour permettre la repolarisation des érythrocytes dans le but de ramener le potentiel membranaire à des valeurs physiologiques. Nous avons utilisé cette stimulation pour dessiner un protocole permettant d’étudier l’impact des conductances cationiques chez les patients piezo1-HX. Ces expériences démontrent que les érythrocytes de patients HX subissent simultanément une déshydratation liée à l’activité aléatoire du canal Gárdos lors d’entrées transitoires et stochastiques de Ca2+ mais également une disruption de l’homéostasie cationique (Na+/K+ ratio), signe marquant du vieillissement accéléré des cellules dans la circulation.
Enfin, nous avons pu utiliser ces protocoles sur des cellules de patients porteurs de mutations non encore décrites Gárdos HSt S314P ; Piezo1 HX D669Y, R2088H et V508M. L’ensemble de ces résultats montrent clairement que les canalopathies du globule rouge sont des pathologies complexes qui incluent le plus souvent une interaction subtile entre le canal Gárdos et le canal Piezo1.
Conclusion. Les caractérisations fonctionnelles de telles mutations sont difficiles dues à l’hétérogénéité des cellules. Nous démontrons que la technique du CCCP est rapide, fiable et peu coûteuse. Elle peut constituer un outil de diagnostic alternatif avec l’avantage de donner des informations sur l’activité des canaux ioniques in situ, corrélées à la sévérité qui peut varier pour des patients porteurs de la même mutation.
19-08
La grille de fragilité short emergency geriatric assessment est utile pour prédire les durées d’hospitalisation longues et les patients justifiant d’un transfert en soins de suite et de réadaptation
D. Chaoui*, N. Al Ayoubi, B. Bertin, L. Hardorock, S. Hebri, V. Mileur, E. Pires, M. Rischebe, S. Seye, E. Vitry, M. Zerzour, S. Touazi, M. Manen, L. Lanier, B. Papoular, B. Kachchour, A. Aljijakli, P. Genet
Hématologie, CH Victor Dupouy, Argenteuil
Introduction. La grille SEGA (pour short emergency geriatric assessment) est un outil de fragilité utile pour prédire les hospitalisations prolongées chez les patients admis aux urgences. Les items sont simples et rapidement recueillis par les infirmières à l’admission des patients. L’objectif de cette étude est d’évaluer l’utilité de cet outil en termes de prédiction de durée d’hospitalisation et du mode de sortie (soins de suite et de réadaptation [SSR] versus domicile) dans un service d’hospitalisation d’hématologie.
Patients et méthodes. De mars 2019 à novembre 2019, la grille SEGA est complétée par les IDE, pour tous les patients de plus de 70 ans. Plusieurs items composent cette grille comme l’âge, les comorbidités, santé psychologique, la qualité de vie actuelle, le nombre de médicaments, les antécédents de chute, les fonctions cognitives, la dénutrition, la perte d’autonomie et l’isolement social. Les patients sont alors classés en trois groupes : un groupe peu fragile (score ≤ 8), un groupe intermédiaire dit fragile (score entre 9 et 11) et un groupe très fragile (score ≥ 12). Pour les patients ayant eu plusieurs séjours d’hospitalisations, seul le premier séjour est retenu pour l’analyse. Les autres informations d’intérêt sont également recueillies : le mode de vie du patient, la date d’entrée et de sortie du service, le type d’hémopathie, le motif d’hospitalisation.
Résultats. Pendant la période d’analyse, 55 patients ont été inclus dans l’étude. L’âge moyen est de 79 ans [70-92]. Les hémopathies sont ainsi réparties : 18 myélomes, 18 lymphomes, huit LLC, six leucémies aiguës, cinq autres hémopathies. Les motifs d’hospitalisations les plus fréquents étaient une prise en charge initiale d’une hémopathie ou une nouvelle phase évolutive d’une hémopathie [n = 33 (60%)], une chimiothérapie programmée [n = 14 (25%)], une complication [n = 6 (11%)]. Environ la moitié des patients (44 %) vivaient seuls. Selon la grille SEGA, 35 patients (64 %) présentaient peu de signes de fragilité. Les autres patients appartenaient soit au groupe fragile (20 %) et très fragile (16 %).
La durée de séjour moyenne était de 12 jours [2-52], significativement différente en fonction du motif d’hospitalisation : chimiothérapie programmée (cinq jours), complications (11 jours), prise en charge d’une phase évolutive d’une hémopathie (19 jours) et en fonction des groupes SEGA : neuf jours pour les groupes peu fragiles et fragiles et 17 jours pour les patients très fragiles.
Concernant le mode de sortie (SSR versus domicile), la majorité des patients très fragiles selon la grille SEGA étaient admis en SSR (75 %) versus 18 % et 19 % pour les patients peu fragiles et fragiles, respectivement.
Conclusion. La grille SEGA est un outil facilement utilisable et utile pour identifier les patients à fort risque de durées d’hospitalisation longues et de transfert en SSR. Cette étude suggère de regrouper les patients peu fragiles et fragiles dans un même groupe.
19-09
Le diagnostic du syndrome d’activation macrophagique secondaire de l’adulte : évaluation des performances du H-Score
I. Hélali1, A. Chakroun*1, S. Mahjoub2, Z. Mannai3, H. Baccouche2, N. Ben Romdhane3
1 Service d’hématologie biologique, CHU la Rabta, Faculté de médecine de Tunis, Tunis, Tunisie ; 2 Service d’hématologie, CHU La Rabta, Tunis, Tunisie ; 3 Laboratoire d’hématologie, CHU La Rabta, Tunis, Tunisie
Introduction. Le syndrome d’activation macrophagique (SAM) est une entité clinique, biologique et cytologique rare pouvant mettre en jeu le pronostic vital. Cette affection est caractérisée par une activation excessive et inappropriée des macrophages et du système lymphoïde à l’origine d’une hémophagocytose incontrôlée. Le diagnostic est un challenge pour le clinicien en raison du polymorphisme de la présentation clinique et l’absence de tests biologiques spécifiques. Le but de ce travail était d’évaluer les performances d’un nouveau score ; le H-Score pour le diagnostic du SAM secondaire de l’adulte en le comparant aux critères HLH-2004 adaptés.
Patients et méthodes. Nous avons mené une étude évaluative prospective au service d’hématologie entre juin 2017 à janvier 2019. Nous avons colligé 54 patients adultes suspects de SAM secondaire Les données épidémiologiques, cliniques, biologiques et cytologiques ont été recueillies. Le HScore ainsi que les critères du HLH-2004 adapté ont été calculés et leurs performances comparées.
Résultats. Les patients ont été répartis en deux groupes : un « groupe SAM », n = 32 et un « groupe non SAM », n = 22. La fièvre, l’hyperferritinémie, l’hypertriglycéridémie et l’élévation de la LDH (p = 0,001, p = 0,001, p = 0,001 et p = 0,004 respectivement) étaient significativement associées au SAM. Le Hscore a montré un excellent pouvoir discriminant entre les deux groupes et était plus performant que le score HLH-2004 adapté (Respectivement, sensibilité = 90,63%, spécificité = 81,82%, VPP = 87,9% et VPN = 85,7% ; AUC = 0,907 pour le H-score versus sensibilité = 78,1%, spécificité = 72,7%, VPP 69,6 % et VPN de 75,93 % ; AUC = 0,825 pour le HLH-2004 adapté.). Les deux scores étaient concordants (indice kappa de Cohen à 0,77).
Dans notre population, le meilleur cut-off du HScore était de 178 permettant gain en spécificité par rapport au cut-off de 169 (sensibilité de 87,5 %, une spécificité de 90,9 %, une VPP de 93,33 % et une VPN de 83,33 %).
Conclusion. Dans notre étude, le Hscore a montré de meilleures performances que les critères du HLH-2004 adapté improprement utilisé chez l’adulte. Ainsi nous encourageons à l’usage du Hscore dans notre pratique clinique. Toutefois, le cut-off adapté à notre population reste à déterminer et à valider par une étude de grande envergure.
19-10
Intérêt diagnostique du séquençage ciblé de l’ARN en hématologie
S. Hayette1, B. Grange1, I. Mosnier1, T. Simonet2, S. Huet1, C. Bardel3, G. Salles4, M. Heiblig5, I. Tigaud1, P. Sujobert*1
1 Service d’hématologie biologique, Hospices civils de Lyon - Hôpital Lyon Sud, Lyon ; 2 Centre de biotechnologies cellulaires, Hospices Civils de Lyon, Lyon ; 3 Bio-Informatique, Hospices Civils de Lyon, Lyon ; 4 Service d’hématologie, CH Lyon Sud, Pierre-Bénite ; 5 Hématologie, CH Lyon-Sud - HCL, Pierre-Bénite
Introduction. Le séquençage du transcriptome (RNA-seq) est une stratégie prometteuse pour améliorer le diagnostic des hémopathies, permettant d’identifier des transcrits de fusion, de rechercher des mutations, et d’analyser le profil d’expression génique. Alors que l’utilisation de cette technologie est devenue habituelle pour la recherche fondamentale, son implémentation en routine diagnostique doit encore faire l’objet d’une évaluation rigoureuse de ses performances analytiques, en particulier des facteurs pré-analytiques qui peuvent impacter les résultats obtenus. Pour répondre à ces questions, nous avons analysé 114 prélèvements diagnostiques et neuf contrôles par RNA-seq ciblé de 1385 gènes impliqués dans le cancer. Nous rapportons ici les performances de cette approche pour identifier des transcrits de fusion, rechercher des mutations somatiques, et mettre en évidence des signatures transcriptomiques spécifiques de certaines hémopathies. Nous décrivons également quels sont les facteurs pré-analytiques qui ont le plus d’impact sur le profil transcriptomique.
Patients et méthodes. 114 prélèvements diagnostiques (51 AML, 28 B-ALL, 23 CML, sept APL, 1 MPAL/AUL, 1 T-ALL, 1 LMMC, 1 SMD, 1 SHE) et neuf témoins ont été inclus. Les prélèvements diagnostiques d’origine médullaire ou sanguine proviennent de patients pris en charge au CH Lyon Sud, et caractérisés au plan cytogénétique et moléculaire. Trois techniques d’extraction d’ARN différentes ont été utilisées (Trizol, Macherey Nagel ou Maxwell) selon les pathologies, et les prélèvements témoins ont été extraits par les trois méthodes. L’analyse bio-informatique a utilisé un pipeline à façon, reposant sur l’utilisation de STAR-Fusion et STAR-2 pass pour la détection des transcrits de fusion, qui ont tous été vérifiés par RT-PCR.
Résultats. Détection des transcrits de fusion : 93 des 94 transcrits de fusion identifiés par les techniques de référence ont été retrouvés, à l’exception d’un cas de réarrangement FIP1L1-PDGFRA. Parmi les 19 prélèvements ayant une translocation chromosomique dont le produit n’était pas caractérisé, nous avons retrouvé un transcrit de fusion déjà décrit dans la littérature pour sept patients (KAT6-CREBP, NPM1-MLF1, PCM1-JAK2, DEK-Nup214, ZMYND11-MBTD1), et de nouveaux transcrits non caractérisés pour six patients (tous validés par RT-PCR). De plus, quatre patients pour lesquels la cytogénétique n’avait pas identifié de translocation avaient un transcrit de fusion. Enfin, par des expériences de dilution, nous avons déterminé la sensibilité de la recherche de transcrit de fusion autour de 5 %.Détection des mutations : Pour 47 patients nous avons pu comparer les performances du RNA-seq pour la détection des mutations somatiques par rapport à l’analyse de l’ADN. 113/129 (87 %) des mutations étaient retrouvées par le RNA-seq, souvent négatif pour la détection des mutations frameshift.Analyse du profil transcriptionnel : Nous avons quantifié l’impact de différents facteurs pré-analytiques sur l’expression des gènes : l’origine tissulaire (sang vs moelle ; cellules mononucléées vs sang total) et la méthode d’extraction ont un impact significatif, alors que l’effet batch n’est pas significatif pour la fabrication ou le séquençage des librairies. En prenant en compte ces facteurs, nous sommes capables de mettre en évidence des profils transcriptionnels significativement différents selon les hémopathies.
Conclusion. Le RNA-seq ciblé est une technique robuste qui permet de préciser les diagnostic des hémopathies, à condition d’en maîtriser les limites pré-analytiques.
19-11
Amélioration du diagnostic moléculaire au travers des évaluations externes de la qualité : 10 années d’expérience du Groupe des biologistes moléculaires des hémopathies malignes
AS. Alary1, C. Maute2, O. Kosmider3, P. Sujobert4, A. Gauthier2, S. Hayette5, D. Luque Paz6, F. Baran-Marszak7, C. Preudhomme8, F. Davi9, E. Macintyre10, E. Lippert11, P. Cornillet-Lefebvre12, MH. Delfau13, B. Cassinat14, JM. Cayuela15, P. Flandrin-Gresta*16
1 Laboratoire d’oncogénétique, Institut Paoli Calmette (IPC), Marseille ; 2 Laboratoire d’hématologie, Hôpital Saint-Louis, Paris ; 3 Hématologie biologique, AP-HP, Hôpital Cochin, Paris ; 4 Hématologie, CH Lyon Sud, Pierre-Bénite ; 5 Biologie moléculaire, CHU, Lyon ; 6 Laboratoire d’hématologie, CHU Angers, Angers ; 7 Laboratoire d’hématologie, Hôpital Avicenne, Bobigny ; 8 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 9 Laboratoire d’hématologie, Hôpital Salpêtrière (AP-HP), paris ; 10 Hématologie, Hôpital Necker, Paris ; 11 Laboratoire d’hématologie, CHU de Brest, Brest ; 12 Biologie, Hôpital Robert Debré, Reims ; 13 Laboratoire d’immunologie et d’hématologie, Hôpital Henri-Mondor AP-HP, Créteil ; 14 Hôpital saint-louis, service de biologie cellulaire, Assistance Publique Hôpitaux de Paris, Paris ; 15 Laboratoire d’hématologie, Hôpital Saint-Louis, Paris ; 16 Laboratoire hématologie, CHU de Saint-Étienne, Saint-Priest-en-Jarez
Introduction. Les tests moléculaires sont aujourd’hui indispensables pour le diagnostic et l’évaluation du pronostic des hémopathies malignes et sont soumis à l’obligation d’accréditation selon la norme européenne ISO 15189. La standardisation de ces techniques et leur confrontation régulière à des évaluations externes de qualité (EEQ) est essentielle. Nous rapportons l’expérience du Groupe des biologistes moléculaires des hémopathies malignes (GBMHM) qui a contribué, par l’organisation des EEQ et de réunions de formation, à l’amélioration et à la standardisation des tests moléculaires de 64 laboratoires incluant la France métropolitaine et les DOM-TOM.
Matériels et méthodes. Une analyse rétrospective des résultats des contrôles qualité de 11 campagnes nationales réparties sur 10 ans (2010 à 2019) a été effectuée pour les trois tests les plus fréquemment prescrits : BCR-ABL, JAK2 et clonalités lymphoïdes.
Résultats. Quantification du gène de fusion BCR-ABL : les laboratoires utilisent majoritairement des méthodes « maison » adaptées du protocole EAC et l’analyse des résultats des EEQ montre une amélioration de la reproductibilité inter-laboratoire. On observe une diminution de la distribution des écarts type (en log) au cours du temps pour les LogRatios BCR-ABL > 0,01 % IS : 0,28 (i.e. 3,6 fois sur les ratios) en 2010 contre 0,14 (i.e. 1,9 fois sur les ratios) en 2019. Cette amélioration permet notamment de réduire l’incertitude des résultats autour de valeurs seuils décisionnelles telle que la réponse moléculaire majeure (RMM). En appliquant les incertitudes de mesure de l’ensemble des laboratoires à une cohorte de 101 patients suivis à Lyon, nous avons estimé que la standardisation avait permis de diviser par 4,7 le nombre de patients dont la RMM aurait été incertaine (6/101 au lieu de 28/101).
Recherche et quantification du gène JAK2 : Les laboratoires utilisent majoritairement des dispositifs de diagnostic in vitro. La reproductibilité inter-laboratoire est satisfaisante et elle a elle aussi évolué favorablement au cours du temps (écarts types à 0,22 en 2010 versus 0,11 en 2019). On observe au cours de ces dix années l’impact des recommandations cliniques sur les pratiques biologiques puisque les tests quantitatifs avec un seuil clinique à 1 % sont désormais utilisés par 88 % des laboratoires, contre 48 % en 2010.
Clonalités lymphoïdes : les laboratoires utilisent de méthodes « maison » adaptées du protocole BIOMED. Grâce aux EEQ mais aussi aux discussions lors des réunions de restitution des EEQ, le GBMHM a contribué à promouvoir une interprétation plus homogène des profils de migration basée sur l’utilisation des recommandations du groupe européen EUROCLONALITY et à l’uniformisation des modalités d’expression des résultats.
Discussion. Ces 10 dernières années, la mise en place des programmes d’EEQ BCR-ABL, JAK2 et clonalités lymphoïdes ainsi que des réunions de formation associées, a permis d’engager un processus de standardisation et d’amélioration des méthodes diagnostiques développées par les laboratoires. Cette standardisation a porté non seulement sur les modalités de mise en œuvre (JAK2) mais aussi sur celles relatives à l’interprétation et à la formulation des résultats (clonalités lymphoïdes). Ce processus a permis d’améliorer de façon objective les performances des laboratoires impliqués (BCR-ABL).
Conclusion. L’activité du GBMHM est donc essentielle pour garantir la meilleure prise en charge des patients et les nouvelles technologies (PCR digitale, NGS), de plus en plus utilisées, doivent également bénéficier de cette stratégie.
19-12
Carence en vitamine B12 et maladies inflammatoires chroniques intestinales : déficit ou association ?
Y. Belattaf*1, S-Mentouri, N-Benfenatki, S-Chemali
1 Médecine interne, hôpital Rouiba, Dar El Beida
Introduction. Les étiologies de la carence en vitamine B12 sont nombreuses et dominées par la maladie de Biermer, maladie auto-immune caractérisée par la présence d’anticorps anti-facteur intrinsèque. La carence en vitamine B12 est également observée au cours des maladies inflammatoires chroniques intestinales (MICI) en raison de l’atteinte iléale ou dans le cadre de l’association d’affections auto-immunes.
Résultats. Nous rapportons cinq observations associant carence en vitamine B12 et MICI :
cas un : patiente âgée de 51 ans, hypertendue, suivie pour RCH colique gauche modérée, diagnostiquée en 2001. En 2011, chez une patiente toujours en rémission sous 5-ASA, découverte sur un bilan de suivi d’une pancytopénie avec anémie sévère 3,6 g/dl macrocytaire mégaloblastique,
cas 2 : patient âgé de 60 ans, sans ATCD particuliers, chez qui une maladie de Crohn colique gauche étendue au transverse inflammatoire en poussée sévère compliquée d’une TVP du MI droit est diagnostiquée en 2018. Au moment du diagnostic, la FNS retrouve une pancytopénie avec anémie sévère 4,1 g/dl macrocytaire mégaloblastique.
Dans les deux cas l’anémie est en rapport avec une carence en vitamine B12 comme le confirme le test à la vitamine B12 revenu positif et une maladie de Biermer. L’aspect endoscopique révèle un aspect d’atrophie fundique dans un seul cas, cependant l’aspect histologique et immunologique est compatible avec une anémie de Biermer dans les deux cas.
Cas (3) : patient âgé de 41 ans, sans ATCD particuliers, suivi pour maladie de crohn ileocolique inflammatoire, diagnostiquée en 2011, en rémission sous 5-ASA. En 2013, deuxième poussée sévère marquée par l’apparition de signes neurologiques, une anémie modérée 9,7 g/dL normocytaire mégaloblastique.
Cas (4) : jeune patient âgé de 27 ans, sans ATCD particuliers, chez qui une maladie de Crohn iléopancolique pénétrante en poussée sévère compliquée d’une TVP du MS droit est diagnostiquée en 2019. Suite à une corticorésistance et sous nutrition parentérale, patient traité par Méthotrexate 20 mg/sem + acide folique et adalimumab 40 mg/2 semaines, il est noté une bonne évolution clinique et paraclinique. Sous traitement, apparition d’une neuropathie périphérique sensitivomotrice confirmée à l’EMG et une légère anémie macrocytaire ; l’IRM médullaire est normale et la recherche d’une néoplasie négative.
Cas (5) : patient âgé de 59 ans, sans ATCD particuliers, chez qui une maladie de Crohn grêlique étagée sténosante est diagnostiquée suite à un syndrome subocclusif. À huit mois de traitement par Azathioprine, une anémie macrocytaire mégaloblastique est apparue.
Dans les trois cas, le test thérapeutique à la vitamine B12 est positif, cependant une maladie de Biermer est éliminée devant l’aspect endoscopique, histologique et immunologique non évocateurs.
Conclusion. La carence en vitamine B12 peut s’observer dans les MICI essentiellement dans la maladie de Crohn par le biais de sa localisation iléale et les conséquences induites (inflammation, médicaments et chirurgie).
Les MICI, maladie de Crohn comme la RCH, peuvent s’associer avec une maladie de Biermer dans le cadre d’associations d’affections auto-immunes.
Il est donc important de comprendre le mécanisme exact de la carence en vitamine B12 et de la corriger. La durée du traitement par la B12 dépendra de l’origine de la carence.
19-13
Prévalence de l’insuffisance rénale chez les patients atteints d’hémopathies malignes, hors myélome multiple, au Maroc : à propos de 766 cas
O. Hari1, EM. Mahtat*1, BH. Mouss1, EM. Hicham1, M. Ababou1, K. Hassani2, K. Doghmi1
1 Hématologie clinique, Hôpital Militaire d’Instruction Mohamed V, Rabat, Maroc ; 2 Néphrologie, hémodialyse et transplantation rénale, Hôpital Militaire d’Instruction Mohamed V, Rabat, Maroc
Introduction. Les données définissant la prévalence de l’insuffisance rénale chez les patients atteints d’hémopathies malignes et son impact sur la prise en charge de ces patients sont relativement rares. Le but de notre travail est de définir la prévalence de l’IR au diagnostic dans les hémopathies malignes hors myélome multiple (MM) prises en charge au Service d’Hématologie Clinique de l’Hôpital Militaire Mohamed V (HMIMV) de Rabat-Maroc et d’évaluer son impact sur la réponse au traitement, le pronostic, la survie globale et sans rechute.
Patients et méthodes. Notre étude est rétrospective, incluant tous les patients suivis pour hémopathie maligne (hors MM) de juin 2006 à août 2019. Les données épidémiologiques, biologiques (taux de créatinine initial et aux dernières nouvelles, taux d’hémoglobine initial, la classification pronostique) et thérapeutiques (prise de médicaments néphrotoxiques, adaptation thérapeutique, réponse au traitement) ont été recueillies à partir de l’archive du service. L’IR était définie par un débit de filtration glomérulaire (DFG calculé selon MDRD) inférieur à 60 mL/min/1,73 m2 et par un taux de créatinine supérieur à 114 μmol/L. L’analyse statistique a été réalisée par le logiciel SPSS.
Résultats. 766 cas d’hémopathies malignes hors MM ont été diagnostiqués sur une période de 13 ans. Le sex-ratio était de 2. La médiane d’âge est de 52 ans. Les hémopathies les plus fréquentes étaient d’origine lymphoïde (69 %), dominées par les lymphomes non hodgkiniens (68 %). 47 % des patients avaient un score de mauvais pronostic. La prévalence de l’IR selon le taux de créatinine était de 7,3 %, et de 11,7 % selon le DFG. Sur le plan thérapeutique, 10 % des patients ont reçu des médicaments néphrotoxiques, répartis entre : les sels de platine (55,26 %), le Méthotrexate (36,84 %) et les anti-inflammatoires non stéroïdiens (7,89 %). Chez les sujets en IR, le protocole a été modifié chez 24 % des patients et les posologies ont été adaptées au DFG chez 51 % des cas. Aux dernières nouvelles, la prévalence de l’IR selon le taux de créatinine était de 10,2 % et de 15,4 % selon le DFG, significativement augmentée par rapport à la prévalence initiale (p = 0,003). Sur un délai de suivi médian de 38 mois, le taux de rechute était de 13,2 % et celui de décès était de 33 %. Nous avons noté une influence significative de la fonction rénale initiale (DFG) sur la réponse au traitement, le taux de RC chez les insuffisants rénaux était de 25 % versus 61,4 % (p < 0,001) chez les patients à fonction rénale normale. Il n’y avait pas de corrélation entre le pronostic de l’hémopathie et la fonction rénale initiale (p = 0,78). La corrélation entre la fonction rénale aux dernières nouvelles et la prise de médicaments néphrotoxiques était non significative (p = 0,69). La médiane de survie globale (SG) et celle de la survie sans rechute (SSR) chez les insuffisants rénaux étaient respectivement de 101 mois et 59 mois (p < 0,001), non atteinte et de 200 mois (p < 0,001) chez les patients sans IR. En analyse multivariée, la présence d’une anémie et d’une insuffisance rénale initiales, l’âge, le score pronostique influençaient significativement la SG et la SSR.
Conclusion. La présence d’une IR au diagnostic d’une hémopathie maligne est fréquente et influence l’évolution de la maladie. Une évaluation prospective et une exploration de l’atteinte rénale initiale chez les patients atteints d’hémopathies maligne est nécessaire pour confirmer les constats de notre étude.
19-14
Étude descriptive et analytique des pancytopénies en milieu hématologique
S. Fall*1, EHD. Niang2, A. Dakono1, S. Mwamba1, NG. Efffa1, FS. Ndiaye2
1 Hématologie cinique, CHN Dalal Jamm, Guédiawaye, Sénégal ; 2 Hématologie clinique, CHN Dalal Jamm, Guédiawaye, Sénégal
Introduction. Les pancytopénies, souvent précédées de bicytopénie, sont caractéristiques par la diversité des étiologies hématologiques et extrahématologiques. Notre objectif était de décrire le profil diagnostique, étiologique et pronostique des pancytopénies.
Patients et méthodes. Il s’agissait d’une étude rétrospective (2016-2019) descriptive, analytique et monocentrique dans un service d’Hématologie Clinique, qui a inclus les dossiers de malades ayant une pancytopénie ou une bicytopénie. Les aspects cliniques, biologiques et le suivi ont été analysés.
Résultats. Ont été recrutés 145 dossiers ; soit une incidence hospitalière de 0,2 % sur trois ans. L’âge moyen était de 48,72 ± 17,31 ans ; et 16,6 % des malades avaient plus 65 ans. Le sex-ratio était de 0,73. Les symptômes étaient un syndrome anémique (71,7 %), un syndrome de réponse inflammatoire systémique (22 %), un syndrome hémorragique (16,5 %), un sepsis (7,6 %) et un cœur anémique (5,5 %). L’hémogramme notait un taux moyen d’hémoglobine de 8,7 ± 2,26 g/dL, de globules blancs de 2,9 ± 0,72 G/L, de polynucléaires neutrophiles de 0,9 ± 0,35 G/L et de plaquettes de 85,6 ± 38,96 G/L. Les signes de gravité biologique étaient présents dans chez 30,3 % des malades.
Les étiologies étaient une aplasie médullaire (41,4 %), une hémopathie maligne (24,2 %), une carence (7,6 %), un hypersplénisme (5,5 %), un syndrome myélodysplasique (3,4 %). Une métastase médullaire (3,4 %), une hémoglobinurie paroxystique nocturne (0,7 %) et le VIH (0,7 %.). La cause n’était pas retrouvée dans 13,1 % des cas.
Sous traitement symptomatique, était notée une correction complète (34,5%), ou partielle (0,7 %) ou une aggravation (10,3 %) des cytopénies. La durée moyenne de suivi était de 556 jours avec une survie médiane à un an de 37 %. L’âge supérieur à 65 ans était corrélé à la survenue de cœur anémique (p = 0,023), des états infectieux (p = 0,041) et de la neutropénie profonde (p = 0,0013).
Conclusion. Les pancytopénies sont des situations nos rares dans nos pratiques et sont d’autant plus graves que le sujet est âgé. Les étiologies sont variées, dominées par l’aplasie médullaire ; d’où la maîtrise d’une démarche diagnostique rigoureuse pour améliorer la prise en charge.
19-15
Étude par cytogénétique conventionnelle et moléculaire des réarrangements 11q23 dans les hémopathies malignes
A. Bennour*, A. Saad, H. Sennana
Cytogénétique, génétique moléculaire et biologie de la reproduction, CHU Farhat Hached, Faculté de médecine, Sousse, Tunisie
Introduction. Les remaniements de la bande chromosomique 11q23 constituent une anomalie de structure fréquente dans les hémopathies malignes, ils conduisent dans la majorité des cas (translocations) à la création d’un gène de fusion entre le gène MLL localisé en 11q23 et un des neuf autres gènes localisés sur les différents chromosomes partenaires. Le produit de fusion présente la structure d’un facteur de transcription impliqué dans la leucémogenèse.
On trouve des translocations de MLL dans la leucémie lymphoïde aiguë (LAL) (3 à 10 %), dans la leucémie myéloïde aiguë (LAM) (8 à 10 %), moins fréquentes dans les syndromes myélodysplasiques (SMD). Leur implication pronostique dépend de l’âge du patient et du phénotype de l’hémopathie. En plus des translocations, des délétions et des amplifications de MLL surviennent également dans un sous-ensemble de LAL et de LAM et SMD.
Du fait de leur pronostic, les réarrangements MLL dans la région 11q23 doivent être recherchés au moment du diagnostic afin d’adapter le traitement en conséquence et de mettre en place une stratégie d’étude de la maladie résiduelle. Ce marqueur peut être défini au niveau cytogénétique mais surtout au niveau moléculaire puisque, comme l’ont montré les techniques moléculaires, un caryotype apparemment normal n’exclut pas un réarrangement de 11q23.
Patients et méthodes. Le but de cette étude est d’évaluer la fréquence des réarrangements 11q23 chez 217 patients diagnostiqués pour différentes hémopathies malignes, en utilisant la cytogénétique conventionnelle (caryotype en bandes R) et moléculaire (FISH avec des sondes spécifiques du gène MLL ; Vysis) ensuite comparer les fréquences révélées par les deux techniques afin de pouvoir discuter leur sensibilité.
Résultats. Les remaniements 11q23 ont été identifiés dans le caryotype de 70 patients (32,25 %). Le réarrangement du gène MLL a été détecté chez 41 patients (18,9 %), ces derniers présentent tous une anomalie 11q23 dans le caryotype. 29 patients (13,3 %) avec des anomalies 11q23 n’avaient pas le réarrangement du gène MLL en utilisant la FISH avec des sondes MLL. Notre étude a confirmé une faible corrélation entre les résultats cytogénétiques conventionnels et moléculaires.
Conclusion. En conclusion, dans le diagnostic des hémopathies malignes, l’analyse cytogénétique conventionnelle seule est nécessaire, mais tout remaniement 11q23 doit être confirmé par la technique moléculaire afin de pouvoir confirmer le réarrangement MLL.
19-16
Évaluation des performances du module d’analyse des liquides biologiques de l’automate Yumizen H2500 (Horiba ®)
A. Inquel1, M. Broly1, M. Chevalier1, K. Viau1, C. Bassi1, MC. Béné1, C. Debord1, C. Chabert2, M. Eveillard*1
1 Service d’hématologie biologique, Hôtel-Dieu, Nantes ; 2 Société horiba Medical, SAS, Montpellier
Introduction. L’analyse automatisée des liquides biologiques est désormais nécessaire pour assurer la robustesse et la fiabilité du rendu des résultats. Ces prélèvements, de nature hétérogène, sont souvent d’interprétation difficile. Aujourd’hui, les automates d’hématologie cellulaire sont en capacité d’analyser certains de ces liquides biologiques. Parmi ces automates, nous avons étudié les performances du module liquide de l’automate Yumizen H2500.
Matériels et méthodes. Une étude prospective a ainsi inclus 100 liquides biologiques pendant 10 semaines. Les prélèvements inclus dans l’étude devaient être en quantité suffisante pour permettre une analyse selon trois modes. En premier lieu, la numération des leucocytes et des érythrocytes était réalisée en cellule de comptage manuelle puis cytospin pour la formule différentielle, le second était un passage en dupliquette sur l’automate Yumizen H2500 et le troisième mode était l’analyse sur l’automate de référence XN10 (Sysmex ®) tant pour la numération des sous-populations (nombre de leucocytes et érythrocytes) que la formule simplifiée (% de cellules mononucléées (%MN) et % de cellules polynucléées (%PN)) ainsi que la détection des cellules anormales. Au final, 98 échantillons ont été analysés selon ces trois modes : 54 liquides synoviaux, 18 liquides pleuraux, 13 liquides de lavage bronchoalévolaire (LBA), six liquides céphalo-rachidien (LCR), cinq liquides d’ascite et deux liquides péricardiques. Les liquides synoviaux nécessitaient un prétraitement par hyaluronidase. Une étape préliminaire de calibration des automates entre eux a été réalisée afin de connaître le % de correction de calibration nécessaire à la corrélation des résultats (3 %). La méthode utilisée pour établir la droite de régression était le Passing Bablok.
Résultats. Corrélation du compte des leucocytes et des érythrocytes : 74 échantillons ont été retenus pour la numération des leucocytes (11 éliminés car situés en dehors du domaine de linéarité des automates et 13 LBA non étudiés car nombreuses alarmes techniques liées à la viscosité des prélèvements). Le coefficient de corrélation entre les deux machines était de 0,991 pour le comptage des leucocytes et de 0,955 entre l’automate YH2500 et la cellule de comptage manuelle. L’étude d’exactitude montre une interférence possible avec la lactescence du prélèvement ainsi que la présence de cristaux sur le nombre de leucocytes. Pour les érythrocytes, le coefficient de corrélation était de 0,996.
Formule partielle : L’étude de la corrélation du % PN entre les deux automates a été possible pour 71 échantillons. Le coefficient de corrélation observé est de 0,933, avec une pente de 0,931 et une intersection à l’origine de +0,18. L’étude d’exactitude montre que la présence de plasmocytes tumoraux dans les prélèvements (n = 3) interfère avec le % PN. Le coefficient de corrélation du % PN entre la formule manuelle et le YH2500 est de 0,914. De la même façon, le coefficient de corrélation du % MN entre les deux machines est de 0,933 et de 0,917 entre la formule manuelle et le YH2500.
Conclusion. Cette étude a permis de tester les performances clinicobiologiques du YH2500 en comparaison à la technique manuelle et la technique de référence automatisée tant pour le comptage des leucocytes et érythrocytes que le différentiel. De plus, elle a permis d’optimiser les seuils de la matrice permettant de différentier les éléments hématologiques des cellules non hématopoïétiques ainsi que d’affiner le positionnement des seuils de différentiation des cellules MN et PN.
19-17
Obinutuzumab en pratique courante : retour d’expérience de 20 patients dans un centre privé
N. Baati*1, A. Iltis-Roux1, V. Graff2, F. Maloisel3
1 Hématologie, clinique Sainte Anne, Strasbourg ; 2 Pharmacie, GHSV, Strasbourg ; 3 Hématologie, Clinique Sainte Anne, Strasbourg
Introduction. Depuis l’obtention de l’AMM dans la leucémie lymphoïde chronique (LLC) en première ligne, chez les patients non éligibles à la fludarabine à pleine dose, puis dans le lymphome folliculaire (LF) en première ligne, l’utilisation de l’obinutuzumab ne cesse de s’étendre. Nous avons évalué la tolérance globale, notamment de la première perfusion et l’efficacité du traitement en pratique courante au sein de notre structure privée.
Résultats. Depuis février 2018, treize patients (pts) ont été traités pour une LLC, en association au Chloraminophène et sept l’ont été pour un lymphome folliculaire, en association avec une chimiothérapie de type CHOP. Pour tous les pts, la prémédication selon les recommandations a été appliquée.
Dans la LLC, huit hommes et cinq femmes ont été traités. L’âge médian est de 77 ans (71-89). Six pts avaient une maladie de stade B évolutif et sept, de stade C. 85 % des pts avaient une lymphocytose supérieure à 25 G/L, 46 % avaient une forte masse tumorale ganglionnaire. Le score de CIRS médian est de 11 (6-20). Six pts avaient une insuffisance rénale chronique avec une clairance de la créatinine inférieure à 50 mL/min. Une réaction à la perfusion est survenue chez six pts (46 %) : une de grade 3 (détresse respiratoire), quatre de grade 2, et une éruption cutanée de grade 1. La patiente ayant présenté la détresse respiratoire avait un antécédent de lobectomie pulmonaire, l’obinutuzumab a été définitivement arrêté. On note la survenue d’un décès par arrêt cardiaque chez une patiente de 76 ans à J5 de la première cure. Son score de CIRS était de 11 avec une insuffisance rénale sévère et une hypertension artérielle traitée. Elle avait présenté une réaction de grade 2 avec hypotension lors du J1. La cause exacte de l’arrêt cardiaque et l’imputabilité du traitement n’ont pu être établies. Un syndrome de lyse tumorale, résolutif, est survenu chez deux pts malgré une prévention systématique. La lymphocytose est normalisée chez tous les pts traités et en vie à J8. Huit pts ont fini leur traitement, un est en échec après six cures. Une neutropénie est survenue dans 30 % des cas dès le premier mois de traitement. Aucune neutropénie fébrile n’est constatée mais trois épisodes infectieux hors neutropénie sont relevés.
Dans le LF : L’âge médian est de 49 ans (26-67). Six pts avaient un stade III/IV. On relève trois réactions de grade 2 à la première perfusion (43%) : une patiente a présenté une douleur abdominale, résolutive sous antalgique. Elle avait un LF rétropéritonéal, stade II avec une forte masse, sans atteinte digestive. Les autres pts ont présenté une hypertension artérielle, des frissons et une sensation d’oppression thoracique sans atteinte cardiaque ni respiratoire. Ces réactions ont été résolutives après arrêt transitoire de la perfusion et traitement symptomatique. Il n’y a eu aucune réaction de grade 3/4 ni aucun syndrome de lyse tumorale. Le traitement a pu être poursuivi chez tous les pts. On relève un seul épisode de neutropénie fébrile malgré un traitement systématique par GCSF, sur un total de 40 cures d’immunochimiothérapie.
Conclusion. Le profil de tolérance de l’obinutuzumab est acceptable en respectant les recommandations de gestion des risques et en hospitalisant les patients pour la première perfusion. Dans la LLC, la sélection des patients éligibles doit être rigoureuse et tenir compte des comorbidités pour limiter l’incidence des effets indésirables graves.
19-18
Épidémie nosocomiale à Burkholderia cepacia dans une unité d’hématologie
B. Achour1, M. Marzouk2, N. Sassi*3, N. Bensayed1, E. Bouslama1, H. Regaieg1, Y. Ben Youssef1, J. Boukadida4, A. Khelif1
1 Hématologie clinique, CHU Farhat Hached, Sousse, Tunisie ; 2 Laboratoire de microbiologie, CHU Farhat Hached,  , Tunisie ; 3 hématologie, CHU Farhat Hached, , Tunisie ; 4 Laboratoire de microbiologie, CHU Farhat Hached, Sousse, Tunisie
, Tunisie ; 3 hématologie, CHU Farhat Hached, , Tunisie ; 4 Laboratoire de microbiologie, CHU Farhat Hached, Sousse, Tunisie
Introduction. Burkholderia Cepacia (BCC) est un bacille à Gram négatif, ubiquitaire, mobile, aérobie, non fermentant. Chez l’homme, cette bactérie multirésistante est une bactérie pathogène opportuniste, responsable d’infections nosocomiales diverses, principalement chez les malades immunodéprimés, ou les bactériémies sont fréquentes.
Matériels et méthodes. Entre février 2018 et août 2018 nous rapportons tous les cas de bactériémies par BCC en milieu d’hématologie chez des patients atteints d’hémopathies malignes et recevant une chimiothérapie intensive.
Résultats. Neuf patients dont deux femmes, d’âge médian de 21,4 ans (de quatre à 45 ans) ont été inclus. Tous étaient sans comorbidités et hospitalisé pour une chimiothérapie pour une hémopathie maligne chez huit patients (LAL : quatre cas, LAM : trois cas, lymphome de Hodgkin : un cas) ou un traitement immunosuppresseur SAL-ciclosporine chez un patient atteint d’une aplasie médullaire. Tous les patients étaient neutropéniques lors de l’infection, avec huit parmi eux ayant une neutropénie grade IV. Le taux moyen de polynucléaires neutrophiles était à 240/μl. Tous les BCC étaient isolés dans les hémocultures et ont montré une sensibilité à l’amikacine et à la ciprofloxacine, et une résistance à la Colimycine. La pipéracilline et le tazobactam n’ont été testés que chez trois cas et tous les BCC y étaient sensibles. Les carbapénèmes ont été testés sur six souches avec une sensibilité à 100 %. Une antibiothérapie initiale appropriée à l’antibiogramme était reçue dans trois cas. Une étude de l’environnement était réalisée et était négative, mais n’a inclus les solutions antiseptiques ou de matériels médicaux.
Pour les scores pronostiques :
le Pitt score était ≥ 2 chez 4 cas
le MASCC score était < 21 chez 3 patients
le Quick sofa était ≥ 2 chez 4 cas
Après une durée d’hospitalisation de 27 jours, on note deux décès en rapport avec l’épisode infectieux.
Conclusion. Les infections causées par Burkholderia cepacia sont fréquemment multirésistantes et peuvent être en cause d’un allongement de la durée d’hospitalisation et d’une mortalité considérable.
19-19
Obinutuzumab (GA 101) : analyse des prescriptions après quatre ans d’utilisation dans un hôpital spécialisé en hématologie
R. Pieragostini*, N. Jourdan-Desrayaud, I. Madelaine-Chambrin, E. Peyrilles
Pharmacie, Hôpital Saint-Louis AP-HP, Paris
Introduction. Le GA-101 est indiqué en association dans le traitement de la leucémie lymphoïde chronique (LLC) et du lymphome folliculaire (LF). Au vu de l’augmentation des prescriptions dans notre hôpital, l’objectif de l’étude était de déterminer la proportion de patients traités par GA-101 conformément aux indications AMM. Les objectifs secondaires étaient d’évaluer la tolérance et l’efficacité de cet anticorps chez les patients traités dans notre établissement.
Patients et méthodes. Une analyse monocentrique rétrospective, de 2016 à 2019, a été conduite à partir des données extraites des dossiers patients informatisés et du logiciel de prescription Chimio®. Tous les patients ayant bénéficié d’au moins une administration de GA-101 ont été inclus.
Résultats. Parmi les 42 patients traités, 69 % (n = 29) l’ont été en accord avec une indication de l’AMM. Hors-AMM, les indications étaient : lymphome B à grandes cellules (n = 2), lymphomes du manteau (n = 1), lymphome B indolent (n = 1), lymphome de Burkitt (n = 1), autre lymphome à petites cellules (n = 1), maladie de Waldenstrom (n = 1) et syndrome de Schnitzler (n = 1). Le GA-101 faisait partie d’une association ne respectant pas l’AMM (vénétoclax, bortézomib + cyclophosphamide) chez deux patients et en monothérapie pour trois patients.
Le traitement d’un LF a concerné 16 patients (38 %) dont 13 (81 %) en stade IV. L’âge médian était 58 ans [33 ; 77]. Le GA-101 était prescrit au moins en deuxième ligne, suite à une progression sous rituximab (RTX) chez 15 patients et suite à un effet indésirable du RTX chez un patient. Les chimiothérapies associées au GA-101 étaient : lénalidomide (n = 9) ou bendamustine (n = 7). Il était bien toléré pour 12 patients. À ce jour, un patient est en rémission complète (RC), neuf patients ont arrêté, après en moyenne 4,6 cycles, pour progression et six sont encore traités.
Treize patients (31 %) présentaient une LLC. L’âge médian de ces patients était 78 ans [34 ; 85]. Le GA-101 a été prescrit en première ligne chez neuf patients (69 %), les autres, avaient tous été précédemment traités par RTX. Il était principalement associé au Chloraminophène (n = 10) ou à l’ibrutinib (n = 2). Seulement huit patients (62 %) ont reçu l’intégralité du traitement (six cures de GA-101). La principale raison d’arrêt était la progression (n = 3). Le GA-101 a été bien toléré chez sept patients (58 %). Une réaction à la première injection ou une cytopénie ont respectivement concerné trois et quatre patients. Sept patients, soit 58 %, sont en RC dont un après progression de la maladie sous RTX. Un patient est encore en cours de traitement.
Un patient parmi les 13 traités hors-AMM n’avait précédemment pas été traité par RTX. Une RC n’a été constatée que chez le patient traité pour un lymphome B indolent.
Conclusion. Dans notre hôpital, le GA-101 est majoritairement prescrit dans le cadre des indications AMM. Concernant la réponse, les résultats dans la LLC sont certes inférieurs à ceux de l’étude pivot mais montrent néanmoins un intérêt du GA-101. Une étude comparant la tolérance et l’efficacité du RTX versus le GA-101 en vie réelle devrait être initiée pour évaluer l’intérêt du GA-101.
19-20
L’impact pronostique des comorbidités au cours des hémopathies malignes du sujet âgé
N. Oubelkacem*, M. Ouazzani, S. Atik, W. Rhandour, I. Khoussar, N. Alami, Z. Khammar, R. Berrady
Médecine interne, CHU Hassan II, Fès, Maroc
Introduction. La fréquence des hémopathies malignes augmente avec l’âge. Les circonstances de découverte sont variables et le pronostic est très souvent défavorable. Le but de notre travail est d’analyser l’impact des comorbidités dans le pronostic, et de définir les différents facteurs pronostic des hémopathies malignes chez la population gériatrique.
Patients et méthodes. Il s’agit d’une étude rétrospective portant sur les sujets âgés de plus de 65 ans pris en charge au service de médecine interne et oncohématologie de CHU Hassan II de Fès, sur une durée de six ans étalée entre 2012 et 2018 et chez qui le diagnostic d’hémopathie maligne a été retenu.
Résultats. Nous avons colligé 148 cas d’hémopathies malignes du sujet âgé, soit une incidence de 24,6 cas/an. L’âge moyen de nos patients était de 73,1 ans ± 6,2, avec des extrêmes de 65 à 95 ans. Le sex-ratio H/F est de 1,05. 38 % de nos patients avaient des comorbidités (diabète, HTA, cardiopathie, etc.). 53% des patients avaient des signes généraux, et 57 % des patients avaient un syndrome inflammatoire biologique.
Les hémopathies étudiées sont : le lymphome non hodgkinien diagnostiqué chez 67 cas (45 % des cas), la leucémie lymphoïde chronique chez 63 cas (42 % des cas), le myélome multiple chez 11 patients (8 % des cas) et la maladie de Hodgkin chez sept cas (5 % des cas).
Une bonne réponse thérapeutique à la première ligne de chimiothérapie est obtenue chez 69 % des patients, 21 patients sont en cours d’évaluation et 14 patients sont perdus de vue. Nous déplorons malheureusement 16 cas de décès (10 %), dont sept cas de LLC, huit cas de lymphome B diffus à grandes cellules et un cas de myélome multiple.
Nous avons trouvé une relation significative entre le décès des patients âgés atteint d’hémopathies malignes et la présence de comorbidités (p = 0,01), la présence de signes généraux (p = 0,04) et la non-réponse à la chimiothérapie de première ligne (p = 0,0003), sans relation ni avec le sexe des patients, ni avec l’âge très avancé (> 75 ans).
Conclusion. Les hémopathies malignes sont fréquentes chez les sujets âgés, surtout le lymphome non hodgkinien et la leucémie lymphoïde chronique. Le décès dans cette tranche d’âge n’est pas dû uniquement à des formes plus agressives mais plutôt à un terrain déjà débilité par une lourde comorbidité.
19-21
Tableau clinicobiologique avec monocytopénie profonde révélant un déficit en GATA 2 : exposé d’un cas présentant la mutation c.1081C>T et mise au point sur le syndrome
Y. Benabed
Laboratoire d’Hématologie, CHU D’Angers, Angers
Introduction. Le déficit en GATA 2 est une maladie constitutionnelle associée à un syndrome d’expression complexe et hétérogène pouvant être caractérisé par une susceptibilité aux infections virales (HPV, EBV), des infections disséminées à mycobactéries non tuberculeuses, un lymphœdème, une monocytopénie profonde et une lymphopénie B et NK. Ces patients ont un risque accru de développer un syndrome myélodysplasique (SMD), une leucémie aiguë myéloïde (LAM) et une leucémie myélomonocytaire chronique (LMMC).
Résultats. Nous rapportons le cas d’une patiente de 23 ans présentant des verrues cutanées planes palmoplantaires, des poussées de panniculite des quatre membres, un érythème noueux, et sur le plan biologique une monocytopénie profonde sans cytopénies associées. Le myélogramme objective une discrète dysmégacaryocytose sans signes francs pour un syndrome myélodysplasique. Une trisomie 8 isolée est retrouvée au caryotype médullaire (47,XX,+8 [7]/46,XX [13]). L’immunophénotypage lymphocytaire met en évidence une lymphopénie B et T CD4+. L’analyse génétique du gène GATA 2 amène finalement à identifier le variant pathogène c.1081C>T (p.Arg361Cys), à l’état hétérozygote. Il s’agit d’une mutation constitutionnelle de novo. L’interrogatoire rapporte un seul antécédent familial chez un de ces quatre frères qui a présenté des infections cutanées et des verrues dans l’enfance. Il est ainsi conclu un déficit en GATA2 avec atteinte cutanée au premier plan. Devant les signes d’hématopoïèse clonale, un projet d’allogreffe est en cours de discussion.
Discussion. Ce variant a été peu rapporté dans la littérature, essentiellement décrit chez plusieurs individus présentant un syndrome myélodysplasique, une monocytopénie et un syndrome MonoMAC (complexe Mycobacterium avium et monocytopénie). Ce variant est classé comme probablement pathogène par le site ClinVar, mentionnant que des données additionnelles sont nécessaires pour l’affirmer formellement.
Conclusion. Cet exposé permet de rapporter ce variant chez une patiente avec un tableau évocateur de déficit en GATA 2.
19-22
Place de la cytologie médullaire dans le diagnostic du syndrome d’activation macrophagique de l’adulte
I. Hélali1, A. Chakroun*1, H. Baccouche2, Z. Mannai3, S. Mahjoub2, N. Ben Romdhane3
1 Service d’hématologie biologique, CHU la Rabta, Faculté de médecine de Tunis, Tunis, Tunisie ; 2 Service d’hématologie, CHU La Rabta, Tunis, Tunisie ; 3 Laboratoire d’hématologie, CHU La Rabta, Tunis, Tunisie
Introduction. Le syndrome d’activation macrophagique (SAM) est une entité clinique, biologique et cytologique rare pouvant mettre en jeu le pronostic vital. Cette affection est caractérisée par une activation excessive et inappropriée des macrophages et du système lymphoïde à l’origine d’une hémophagocytose non contrôlée. Le diagnostic est problématique en raison du polymorphisme des signes cliniques et l’absence de test biologique spécifique. L’image d’hémophagocytose n’est ni spécifique, ni constante au cours du SAM. L’objectif de notre étude était de rechercher des critères cytologiques permettant d’optimiser l’orientation diagnostique.
Patients et méthodes. Nous avons mené une étude prospective évaluative monocentrique ayant colligé tous les cas de suspicion d’un syndrome d’activation macrophagique (SAM/HLH secondaire) de l’adulte présentés à notre service d’hématologie afin de réaliser un myélogramme et ce dans la période allant de juin 2017 à janvier 2019. Ont été inclus tous les cas adultes, âgés de plus de 16 ans, chez qui le diagnostic de SAM a été suspecté par l’équipe médicale en charge du patient. Les données épidémiologiques, cliniques, biologiques et cytologiques ont été recueillies. Une deuxième lecture des frottis médullaires a été réalisée en aveugle. Le nombre de macrophages/1 000 éléments nucléés, L’intensité de l’hémophagocytose, la nature des éléments phagocytés ainsi que la présence d’images de phagocytose multiple ont été précisés.
Résultats. Nous avons colligé 54 suspicions de SAM. Au final, 35 frottis médullaires ont été étudiés. Deux groupes de patients ont été constitués sur la base du diagnostic définitif retenu par l’équipe médicale : Un « groupe SAM », n = 24 et un « groupe non SAM », n = 11. Les paramètres associés au diagnostic de SAM étaient la fièvre, la ferritinémie, la triglycéridémie et LDH (respectivement, p = 0,007 ; p = 0,04 ; p = 0,000 ; p = 0,034). En revanche aucun des paramètres cytologiques étudiés à la deuxième lecture n’était associé au diagnostic de SAM. Nous avons procédé à une étude de régression multiple intégrant des paramètres clinicobiologiques classiquement introduits dans les critères diagnostiques, mais également des critères de l’étude cytologiques. Les trois modèles établis avaient des capacités moyennes de prédiction du diagnostic de SAM. La contribution des nouveaux critères cytologiques ne semble pas améliorer la qualité des équations.
Conclusion. Une étude médullaire sur 1 000 éléments nucléés, qui peut être longue et fastidieuse, se heurte avec le caractère urgent du diagnostic. Néanmoins, des guidelines concernant la lecture du myélogramme chez les patients suspects de SAM restent à définir.
19-23
Complications neurologiques sévères au méthotrexate haute dose au cours du traitement des leucémies aiguës lymphoblastiques
N. Ben Sayed*1, W. Chambeh2, B. Achour3, E. Bouslama3, R. Haifa3, YY. Ben3, A. Khelif3
1 Service d’hématologie clinique, CHU Farhat Hached, Sousse, Tunisie ; 2 Service d’hématologie clinique, CHU Farhat Hached, , Tunisie ; 3 Hématologie clinique, CHU Farhat Hached, Sousse, Tunisie
Introduction. Les leucémies aiguës lymphoblastiques aiguës (LAL) sont des hémopathies malignes très fréquentes chez l’enfant. Le traitement se base sur la polychimiothérapie associant plusieurs drogues ainsi les stratégies d’intensification des doses ont entraîné une amélioration significative des survies. Les antagonistes du folate tels que le méthotrexate (MTX) ont été parmi les premiers agents antinéoplasiques à être développés. Cette molécule est un anticancéreux de la famille des cytostatiques qui traversent la barrière hémato-encéphalique donc peut passer dans le LCR pour pouvoir neutraliser le maximum de cellules cancéreuses. En effet son administration à des fortes doses permet de vaincre les phénomènes de résistance cellulaire mais peut provoquer des toxicités sévères parfois létales. Aussi la réduction de dose peut être inefficace réduisant les chances de réponse au traitement.
Patients et méthodes. Il s’agit d’une étude descriptive rétrospective réalisée dans le service d’hématologie clinique de l’hôpital Farhat Hached de Sousse, sur une période de dix ans étendu du 1er janvier 2008 jusqu’au 31 décembre 2017. Nous avons colligé les observations de 181 patients hospitalisés au service d’hématologie clinique atteint de LAL traité par une polychimiothérapie contenant le méthotrexate et qui ont développé une toxicité liée à cette drogue.
Résultats. Au cours de l’évaluation des données, quatre épisodes de toxicité neurologique grave ont été identifiés soit 2,2 % de l’ensemble des toxicités. Il s’agit de deux filles et deux garçons dont l’âge moyen est de 14 ans, tous atteints de leucémie aiguë lymphoblastique : de type B chez trois patients et de phénotype T chez un seul patient. Deux patient ont reçu une chimiothérapie selon EORTC VHR, 1 patient selon le protocole hyper-CVAD et une selon lez protocole Saint Jude VHR. En effet les patients ont présenté, après l’injection du MTX haute dose (HD), des troubles neurologiques différents à type de troubles du comportement, de confusion mentale et de crises convulsives tonicocloniques chez deux patients. Cette symptomatologie est apparue après un délai moyen de 76 jours du début du traitement (35-120 j). Les quatre patients ont eu des IRM cérébrales montrant un aspect de PRESS sd chez deux patients et un aspect de leucoencéphalite toxique chez un patient. Le sauvetage folinique était donné à double dose chez tous les patients, avec une bonne évolution chez trois patients et résolution progressive des troubles neurologiques et aggravation chez un patient. Seulement une seule patiente a reçu de nouveau le MTX HD de nouveau à distance de l’épisode confusionnel sans récidive de la symptomatologie neurologique. Trois patients sont décédés suite à une progression de leur maladie et une seule patiente est actuellement en rémission avec un recul de 56 mois.
Conclusion. Le MTX constitue une drogue maîtresse dans la prise en charge curative des LAL, à dose usuelle ou à forte dose. L’utilisation de cette dernière nécessite l’adaptation de sa posologie et le sauvetage folinique afin d’éviter les toxicités.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License