Hématologie
MENU17 Immunohématologie Volume 26, supplement 4, Septembre 2020

17-01
Combinaison d’agents thérapeutiques pour la prise en charge de la leucémie dérivée de cellules dendritiques plasmacytoïdes
M. Poussard*, E. Bole-Richard, M. Fredon, S. Biichle, P. Saas, O. Adotevi, F. Garnache Ottou, F. Angelot-Delettre
TIM-C, UMR1098, EFS BFC, Université de Franche-Comté, Besançon
Introduction. La leucémie à cellules dendritiques plasmacytoïde (LpDC) est une hémopathie rare et très agressive pour laquelle aucun consensus de traitement n’est actuellement établi. La LpDC semble chimiosensible puisque deux patients sur trois répondent aux chimiothérapies standards, mais des rechutes rapides surviennent fréquemment avec le développement de chimiorésistance. Seuls les patients jeunes, ayant pu bénéficier d’une polychimiothérapie suivie d’une intensification par allogreffe de cellules souches hématopoïétiques (CSH) en première rémission complète, obtiennent des rémissions à plus long terme. Suite à une étude rétrospective de plusieurs cas de patients, quatre chimiothérapies ont présenté un intérêt lorsqu’elles sont utilisées en combinaison. Cette dernière, nommée LpDessai fait actuellement l’objet d’un essai clinique de phase II au CHRU de Besançon, évaluant son efficacité chez des patients atteints de LpDC. Cependant, les rechutes étant fréquentes et l’allogreffe de CSH, une thérapie lourde et accessible à peu de patients âgés, nous avons souhaité évaluer l’efficacité d’une combinaison de polychimiothérapie et d’immunothérapie comme alternative à la greffe de CSH. Pour cela nous avons modélisé in vivo cette association dans un modèle murin de LpDC en associant la combinaison LpDessai et l’immunothérapie de type CAR-T cells (lymphocytes T génétiquement modifiés pour exprimer un récepteur chimérique à un antigène (CAR)) ciblant le CD123, un cluster de différenciation fortement exprimé dans la majorité des cas de LpDC.
Matériels et méthodes. Les lignées cellulaires CAL-1 et GEN2.2, dérivées de patients atteints de LpDC, ainsi que des cellules primaires de huit patients LpDC ont été traitées avec les quatre chimiothérapies seules ou en association. La viabilité cellulaire a été évaluée après 24 h de traitement par cytométrie en flux (CMF). Des souris immunodéficientes NSG-S ayant reçu une injection d’une lignée cellulaire de LpDC (CAL-1 luciférase) ont été utilisées comme modèle murin de LpDC et traitées à J2 par la combinaison LpDessai. A J4, les souris traitées par la polychimiothérapie ont été traitées par les LT CAR CD123. Des groupes traités par la polychimiothérapie seule, des lymphocytes T non modifiés seuls ou des LT CAR CD123 cells seuls ont été utilisés comme contrôle. La progression tumorale a été suivie chaque semaine par bioluminescence. Les LT CAR CD123 cells ont été produits par transduction de LT de donneurs sains avec un lentivirus. L’efficacité de transduction et la cytotoxicité ont été évaluées par CMF.
Résultats. L’efficacité de transduction des LT CAR CD123 est élevée (98,4 % à J12) et stable au cours du temps. Leur cytotoxicité est importante et spécifique face à leurs cibles (99,9 % de cytotoxicité sur des cibles CD123+). In vivo, la polychimiothérapie prolonge faiblement la survie mais lorsqu’elle est utilisée en association avec les LT CAR CD123 la progression tumorale est contrôlée et la survie des souris est significativement prolongée par rapport au groupe non-traité. Le groupe traité avec les CD123 CAR-T cells seuls montre les mêmes résultats. Ainsi les chimiothérapies n’impactent pas la fonctionnalité des LT CAR CD123.
Conclusion. Nous rapportons in vivo l’intérêt d’utiliser une thérapie de type CAR-T cells en seconde ligne de traitement suite à une polychimiohtérapie pour le traitement de la LpDC. En effet, cette dernière ne semble pas impacter l’effet de la thérapie cellulaire. Les CAR-T cells pourraient donc se présenter comme une alternative à la greffe de CSH, très agressive et donc peu accessible aux personnes atteintes de LpDC, majoritairement âgées.
17-02
Angioedèmes par déficit acquis en C1-inhibiteur et hémopathies
D. Gobert*1, G. Kalmi1, F. Defendi2, G. Hardy3, V. Frémeaux-Bacchi4, N. Javaud5, V. Panayatopoulos1, L. Bouillet6, D. Roos-Weil7, R. Paule8, P. Coppo9, O. Fain10, CREAK
1 Médecine interne, Hôpital Saint-Antoine AP-HP, Paris ; 2 Laboratoire d’immunologie, CHU Grenoble Alpes, Grenoble ; 3 Um génétique moléculaire : maladies héréditaires et oncologie, CHU Grenoble Alpes, Grenoble ; 4 Service d’immunologie biologique, inserm unité 872, Hôpital Européen Georges-Pompidou AP-HP, Paris ; 5 Urgences, Hôpital Louis Mourier, Colombes ; 6 Médecine interne, CHU Grenoble Alpes, Grenoble ; 7 Hématologie, Pitié Salpêtrière, Paris ; 8 Médecine interne, hôpital Foch, Suresnes ; 9 Hématologie clinique, CHU Saint-Antoine, Paris ; 10 Médecine interne, hôpital Saint-Antoine, PARIS
Introduction. Les angiœdèmes bradykiniques par déficit acquis en C1-inhibiteur sont une entité rare, souvent associée à des lymphomes – notamment de la zone marginale – et des gammapathies monoclonales. Les épisodes d’angiœdème surviennent de manière imprévisible, et peuvent occasionner des atteintes laryngées engageant le pronostic vital, et des douleurs abdominales intenses.
Patients et méthodes. Nous avons conduit une étude nationale rétrospective des cas d’angiœdème par déficit acquis en C1-inhibiteur en France depuis 1991 jusqu’à 2019, signalés via le réseau de centres de référence des angiœdèmes à kinine (CREAK) (N = 28 sites) et des laboratoires d’immunologie de référence de l’hôpital de Grenoble et de l’hôpital européen Georges Pompidou, à Paris.
Résultats. Nous avons recueilli les données concernant 121 patients, dont 69 femmes, d’âge médian de 63 ans [33-89] au début des symptômes. Les épisodes d’angiœdème atteignaient la face (76 %), l’abdomen (60 %), la langue (31 %) et le larynx (41 %), et survenaient en moyenne une fois par mois (allant d’une fréquence hebdomadaire à quelques crises par an selon les individus). Le diagnostic d’angiœdème bradykinique était porté après un délai moyen de 1,7 an après l’apparition des œdèmes. Une hémopathie lymphoïde était présente chez 50 % des patients : lymphomes de la zone marginale (n = 29), lymphomes B inclassés (n = 10), leucémies lymphoïdes chroniques (n = 8), maladies de Waldenström (n = 7), lymphomes lymphocytiques (n = 3), lymphomes B diffus à grandes cellules (n = 2), un lymphome de type grey-zone et un lymphome folliculaire. Une gammapathie monoclonale isolée était observée chez 28 % des patients, d’isotype IgM (n = 17), IgG (n = 11) et IgA (n = 6). L’angioedème précédait le diagnostic d’hémopathie chez 54 patients avec un délai moyen de trois ans [0,6-16], et était contemporain du diagnostic chez 22 patients. Un anticorps anti-C1-inhibiteur était présent chez 46 % des sujetqs, ne correspondant pas systématiquement à l’isotype du pic monoclonal identifié. La réponse aux traitements d’urgence des crises (icatibant, concentrés de C1-inhibiteur) était proche de 100 %. Un traitement spécifique de l’hémopathie a été introduit chez 49 patients (80 %) : splénectomie (n = 7), chimiothérapie (n = 30), et rituximab ± chimiothérapie (n = 55) et a permis une rémission clinique des épisodes d’angiœdèmes dans 100 %, 70 % et 78 % des cas respectivement. Après un suivi moyen de 6,2 ans (0-21 ans), sept patients étaient décédés, dont un d’un angiœdème laryngé.
Conclusion. Cette entité est méconnue, conduisant à une errance diagnostique, notamment devant des épisodes douloureux abdominaux attribués à tort à la splénomégalie. La répétition des crises d’angiœdème bradykinique et leur gravité potentielle peut justifier le traitement d’hémopathies parfois indolentes. La place de nouveaux traitements ciblés tels que les inhibiteurs de la voie du BCR et les anti-BCL2 doit être évaluée dans cette indication. Les patients doivent toujours disposer d’un traitement d’urgence des crises, dont l’efficacité est comparable à celle observée dans la forme héréditaire.
17-03
Mécanismes de clairance érythrocytaire durant l’anémie hémolytique liée au traitement du paludisme grave
C. Chambrion*1, M. Depond2, EF. Gautier3, O. Mouri4, M. Dussiot5, E. Kendjo4, FM. Aurélie1, C. Roussel6, T. Aida1, N. Argy7, S. Houze7, P. Mayeux3, R. Piarroux8, JY. Siriez9, S. Larréché10, M. Thellier4, P. Buffet1, PA. Ndour1, Groupe de travail sur l’Artesunate
1 Umr-s1134, Institut National de la Transfusion Sanguine, Paris ; 2 Ints, Inserm U1134, Paris ; 3 Inserm U1016, CNRS UMR 8104, université paris Descartes, Labex GR-EX, Institut Cochin, Paris ; 4 Cimi-paris u1135, Hôpital Pitié Salpêtrière Pavillon Lavéran, Paris ; 5 Grex, Université Paris Descartes, Paris ; 6 Umr-s 1134, Institut National de la Transfusion Sanguine (INTS), Paris ; 7 Centre national de référence du paludisme, Hôpital Xavier Bichat-Claude Bernard, Paris ; 8 Centre national de référence du paludisme, Hôpital Pitié Salpêtrière Pavillon Lavéran, Paris ; 9 Pédiatrie, Hôpital Robert-Debré AP-HP, Paris ; 10 Biologie médicale, Hôpital d’instruction des armées Bégin, Paris
Introduction. Les dérivés de l’artémisinine sont recommandés en première intention par l’OMS pour traiter le paludisme grave. Ces médicaments tuent les stades jeunes du parasite, prévenant ainsi la séquestration des globules rouges parasités dans les capillaires. Le parasite mort est ensuite expulsé du globule rouge lors de son passage dans les fentes inter-endothéliales de la rate. Une fois débarrassé de son parasite, le globule rouge dit « pitté » est remis en circulation. Une anémie hémolytique différée (PADH) liée à la destruction des globules rouges pittés est observée environ dix jours après le début du traitement. Cet effet secondaire concerne 5 à 25 % des sujets traités. Deux protéines parasitaires laissées en tant qu’empreintes après le pitting, RESA et HRP2, permettent aujourd’hui de prédire la PADH par quantification des globules rouges pittés. Toutefois, durant la PADH, la perte de globules rouges est supérieure à la totalité des globules rouges pittés. La caractérisation des globules rouges éliminés durant la PADH vise à mieux comprendre la pathogenèse de cette nouvelle anémie hémolytique.
Patients et méthodes. Nous avons étudié des globules rouges de patients traités par dérivés de l’artémisinine avant et après un épisode de PADH, ainsi que des globules rouges pittés générés ex vivo dans les rates isolées perfusées. L’identification de marqueurs des globules rouges modifiés a été réalisée à l’aide d’une approche protéomique par spectrométrie de masse.
Résultats. Nous avons observé que la quantité de protéines parasitaires et la proportion de globules rouges pittés présents dans les échantillons étaient corrélées. Nous avons également noté une persistance de plus d’une centaine de protéines parasitaires dans les globules rouges, dont 20 potentiels marqueurs de clairance, qui disparaissaient donc de la circulation durant la PADH. Certaines d’entre elles sont connues pour être associées à des phénomènes d’anémies, comme Rhoptry-Associated Protein-2, et d’autres à la déformabilité du globule rouge parasité, comme la Knobs-Associated Histidin-Rich Protein.
L’étude par imagerie en flux des globules rouges pittés de patients ou générés ex vivo a montré qu’ils sont plus petits que les globules rouges sains. Cette réduction de surface pourrait expliquer la capacité réduite des globules rouges pittés à franchir la barrière splénique. Par ailleurs, les analyses en spectrométrie de masse montrent une augmentation de la fixation des Immunoglobulines à la surface des globules rouges et une diminution de l’expression de protéines inhibitrices de la lyse par le complément juste avant et pendant la PADH.
Conclusion. Ces résultats suggèrent que l’élimination des globules rouges durant la PADH peut s’expliquer à la fois par une diminution de la déformabilité des globules rouges pittés, induisant leur rétention mécanique dans la rate, une reconnaissance immune spécifique (fixation d’immunoglobulines et activation du complément) des globules rouges pittés, ou modifiés lors de l’infection indépendamment du pitting, les deux pouvant contribuer à l’anémie. Ce mécanisme d’amplification de l’anémie pourrait intervenir dans d’autres anémie hémolytiques.
17-04
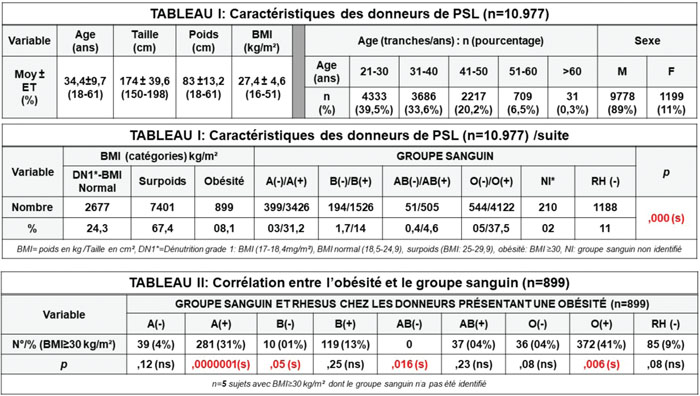
Est qu’il existe une corrélation entre le groupe sanguin érythrocytaire et la tendance à l’obésité à propos d’une cohorte de 10 977 donneurs de sang de la région de Boufarik ?
F. Talbi*1, D. Damerdji2, R. Nebbab3
1 hématologie, hôpital Central De l’Armée DR Mohamed Seghir Nekkache, Alger, Algérie ; 2 Poste de transfusion sanguine, EPH de Boufarik, Boufarik, Algérie ; 3 Épidémiologie, Hôpital Mayo, Alger Centre, Algérie
Introduction. Nous avons évalué la prévalence de l’obésité, en fonction du groupe sanguin, sur une cohorte de 10 977 donneurs de sang de la région de Boufarik.
Matériels et méthodes. Il s’agit d’une étude ambidirectionnelle sur 11 ans. Les données sont exportées en Excel par Epidata d’un fichier DBF (logiciel auto-exécutable) réalisé sous Cliipper de gestion d’un centre de TS. N = 10 977 dons de sang issus de huit établissements sont enregistrés pendant la période d’étude. Les réactifs Bio-rad® sont utilisés pour le groupage ABO, RH. La taille est mesurée en cm et le poids en kg, le BMI (body mass index) est calculé par la formule standard : poids (kg)/taille2 (cm) : un poids normal = BMI (18,5-24,9 kg/m2), le surpoids : BMI (25-29,9 kg/m2) et l’obésité (BMI ≥ 30 kg/m2). Les résultats sont exprimés sous forme de moyennes ± écart-type, l’association du BMI et les groupes sanguins est évaluée par analyse de la régression. Une valeur p < 0,05 est jugée statistiquement significative.
Résultats. N = 10 977 donneurs de PSL sont répertoriés de 2008 à 2018), n = 9778 H/1199 F ; sex-ratio = 8,15, (les caractéristiques de notre population d’étude sont présentées dans les tableaux 1 et 2), en moyenne : l’âge = 34,4 ans, le poids = 83 kg, la taille = 174 cm, le BMI = 27,4 kg/m2; n = 899 (08,1 %) présentent une obésité. Le groupe O = 42,5% de notre effectif. La corrélation entre le groupe ABO et le BMI : p < 0,000. L’analyse du risque d’obésité selon le groupe sanguin montre une corrélation robuste avec : le groupe A(+): p = 10-7, O(+): HR = 1,4, p = 0,006, AB (-) : p = 0,016 (HR = 9,1) et le groupe B (-) (p à la limite de la significativité : =,05).

Conclusion. Il ressort de notre étude que le groupe(ABO) affecte considérablement les probabilités d’être en surpoids ou obèse, cette prédisposition concerne les groupes : A(+), 0(+) et, à degrés moindre, le groupe B(-). Le groupe AB négatif présente un effet protecteur sur les risques de surpoids (et d’obésité).Jawed S a rapporté des données similaires en ce qui concerne la corrélation positive entre l’obésité et le groupe O et l’effet protecteur du groupe AB, cependant, dans notre cohorte, cet effet (protecteur) se limite au RH négatif (AB-).
17-05
Quelle gestion des anti-thrombotiques pour les embolies pulmonaires au cours des purpura thrombotique thrombocytopénique auto-immuns traités par caplacizumab ?
R. Cauchois*1, PA. Jarrot1, F. Sanderson2, K. Mazodier1, R. Jean1, P. Suchon3, R. Lacroix4, E. Tellier5, F. Dignat-George5, G. Kaplanski1, P. Poullin2
1 Médecine Interne et Immunologie clinique, Hôpital de la Conception, Marseille ; 2 Hémaphérèse et autotransfusion, Hôpital de la Conception, Marseille ; 3 Hématologie biologique, CHU Timone, Marseille ; 4 Service d’hématologie et de biologie vasculaire, CHU Conception, APHM, Marseille ; 5 Inserm, inrae, c2vn, Aix-Marseille Université, Marseille
Introduction. Le caplacizumab est le premier médicament à obtenir une autorisation de mise sur le marché pour le traitement du purpura thrombotique thrombocytopénique acquis (PTT-a) en association aux échanges plasmatiques et aux immunosuppresseurs et à poursuivre au moins 30 jours après l’arrêt des échanges. Il s’agit d’un nanobody qui inhibe l’interaction entre le domaine A1 du Facteur von Willebrand et le Gp1b plaquettaire, conférant aux patients un phénotype de maladie de Willebrand de type 2M, donc potentiellement hémorragique. Nous rapportons trois cas d’embolies pulmonaires (EP) survenus dans notre cohorte de 13 patients traités par caplacizumab depuis août 2018.
Résultats. Trois femmes âgées de 35, 59 et 70 ans, ont présenté une poussée de PTT-a traitée par échanges plasmatiques, corticothérapie (1 mg/kg), caplacizumab (dès J1) et rituximab (dès J2). Deux patientes ont été initialement admises en réanimation pour des atteintes cardiaques ou neurologique, les échanges plasmatiques étant alors réalisés sur cathéter fémoral. Aucune patiente n’a bénéficié d’une anticoagulation préventive compte tenu de la thrombopénie majeure (< 20 G/L). Chez un patiente, l’EP segmentaire apicale droite a été diagnostiquée à J5 sur un angio-TDM thoracique réalisé pour des douleurs thoraciques, tandis que chez les deux autres, les diagnostics d’EP bilatérales étaient portés fortuitement à J7 et J8 sur des angio-TDM thoraciques réalisés à la recherche d’une néoplasie sous-jacente. Dans un cas, l’EP était associée à un caillot de 5 cm dans le cathéter fémoral. Toutes les patientes ont été traitées par enoxaparine à dose efficace adaptée au poids, relayée par warfarine (n = 1) ou apixaban (n = 2) après arrêt du caplacizumab. Une patiente a présenté de volumineux hématomes douloureux de l’abdomen aux points d’injection nécessitant l’arrêt précoce du caplacizumab (J21/30) alors que l’activité ADAMTS13 était normalisée.
Discussion. Ces trois observations illustrent les difficultés de diagnostic et de traitement de l’EP des patients atteints de PTT-a traités par caplacizumab. L’EP est une complication probablement sous-diagnostiquée au cours du PTT : il existe en effet de nombreux facteurs favorisant (immobilisation en réanimation, fréquence des cathéters centraux, échanges plasmatiques), et la symptomatologie aspécifique de l’EP peut être facilement attribuée à l’anémie ou à l’ischémie viscérale. Dans notre expérience, la réalisation d’angio-TDM systématiques a permis de diagnostiquer deux EP asymptomatiques chez trois de nos patientes. À la phase initiale du PTT, l’utilisation d’une anticoagulation préventive est rare car les patients ont un risque hémorragique théorique élevé, lié à la profonde thrombopénie et au mécanisme d’action du caplacizumab. Nous proposons désormais qu’une anticoagulation préventive soit instaurée dès le taux de plaquettes > 50 G/L. Sous caplacizumab, le choix du traitement de l’EP est délicat. Étant donné le risque hémorragique élevé, nous avons opté pour une HBPM à courte demi-vie, selon une posologie adaptée au poids. La survenue de saignements doit faire discuter l’arrêt prématuré du caplacizumab. Des données de pharmacovigilance supplémentaires sont nécessaires concernant une éventuelle imputabilité du caplacizumab dans la maladie thromboembolique veineuse.
Conclusion. L’EP est une complication probablement sous-diagnostiquée au cours du PTT. La balance bénéfice risque d’une anticoagulation préventive à la phase aiguë et la gestion de l’anticoagulation efficace en association au caplacizumab doivent être évaluées.
17-06
Atteinte hématologique au cours du lupus érythémateux systémique : analyse d’une série de 440 cas
W. Chawad, W. Ammouri*, Z. El Hazaz, H. Khibri, M. Bouaouad, M. Maamar, H. Harmouche, Z. Tazi Mezalek, M. Adnaoui
Service de médecine interne/hématologie clinique, CHU IBN Sina, Faculté de médecine et de pharmacie, Université Mohammed V, Rabat, Maroc
Introduction. Le lupus érythémateux disséminé (LED) représente l’archétype des maladies auto-immunes avec une implication multisystémique variable et des caractéristiques cliniques hétérogènes. Les anomalies hématologiques sont fréquentes et font parties des critères diagnostiques chez les patients atteints de LED. Elles se manifestent surtout par des cytopénies, pouvant toucher toutes les lignées. Isolées ou associées aux autres manifestations, elles ont habituellement peu de traduction clinique et ne nécessitent que rarement un traitement spécifique. La splénomégalie et les adénopathies périphériques sont souvent contemporaines d’une poussée de la maladie. D’autres atteintes hématologiques graves peuvent mettre en jeu à court terme le pronostic vital tels que la lymphohistiocytose hémophagocytaire, la microangiopathie thrombotique et de façon exceptionnelle, la myélofibrose.
Patients et méthodes. Il s’agit d’une étude rétrospective monocentrique de type descriptive et analytique réalisée sur des patients marocains hospitalisés et pris en charge au service de médecine interne du CHU Ibn Sina de Rabat, pour un lupus érythémateux disséminé, sur une période de 24 ans (de janvier 1990 à janvier 2014). Cette étude a inclus des patients âgés de plus de 15 ans, répondant aux critères modifiés de L’ACR. L’objectif de notre étude était de décrire les aspects épidémiologiques, cliniques, et paracliniques de l’atteinte hématologique au cours du LES.
Résultats. Au total, 440 cas ont été colligés, dont 393,8 femmes (soit 89,5 %) et 6,2 hommes (10,5 %) soit un sex-ratio F/H de 63,51. L’âge moyen était de 32 ans avec des extrêmes allant de 20 à 43 ans. L’atteinte hématologique était présente chez 85 % des patients (soit chez plus des 2/3). 20 % Des patients avaient une splénomégalie associée à des adénopathies périphériques concomitantes à une phase d’activité de la maladie. La leucopénie a été retrouvée chez 176 (40 %) patients. Une baisse du taux de lymphocytes était notée dans 70 % des cas. L’anémie a été notée dans 40 % des cas à type d’anémie hémolytique auto-immune à autoanticorps chauds. Quant à la thrombopénie, elle a été notée dans 30 % des cas et n’a entraîné aucune manifestation hémorragique grave. Parmi les manifestations hématologiques graves, un syndrome d’activation macrophagique a été recensé chez 20 patients (soit 4,5 %). Il était contemporain d’une poussée lupique chez 13 patients et associé à une infection dans 13 cas. L’atteinte hématologique était révélatrice de la maladie lupique chez 80 % des patients, pour les autres elle était associée aux poussées de la maladie. En analyse univariée, les facteurs significativement associés à l’atteinte hématologique étaient : l’atteinte rénale, les ulcérations buccales, le rash malaire, l’atteinte cutanée, la présence d’anticoprs antiphospholipides, les anticorps anti-SS A et SSB, les anti-Sm et une forme grave de lupus (p < 0,05).
Conclusion. L’atteinte hématologique est fréquente au cours du lupus et doit être recherchée avec soin. Elle représente souvent le mode de révélation de la maladie avec des mécanismes variés. En pratique, il est important de distinguer une atteinte hématologique spécifique de la maladie d’une atteinte iatrogène médicamenteuse, et de reconnaitre rapidement les atteintes pouvant engager à court terme le pronostic vital.
17-07
Particularités du syndrome d’activation macrophagique en milieu de médecine interne
F. Abida1, R. Ben Salah*2, I. Chabchoub2, M. Châari1, B. Zouhir2
1 Laboratoire d’hématologie, CHU Hédi Chaker, Sfax, Tunisie ; 2 Médecine interne, CHU Hédi Chaker, Sfax, Tunisie
Introduction. Le syndrome d’activation macrophagique (SAM) est une pathologie rare et potentiellement létale. Il peut être primaire ou secondaire. Son diagnostic peut être difficile et doit amener à la réalisation d’une enquête étiologique précise. Le but de ce travail est de préciser les caractéristiques cliniques, biologiques, étiologiques et évolutives des cas de SAM dans un service de médecine interne.
Patients et méthodes. Étude descriptive rétrospective réalisée sur une période de 22 ans (1996-2018) incluant les dossiers des patients hospitalisés dans un service de médecine interne et présentant un SAM dont le diagnostic était retenu selon les critères d’Histiocyte Society 2004.
Résultats. Il s’agissait de 10 patients : huit femmes et deux hommes d’âge moyen de 47,2 ans. Les patients présentent des antécédents d’un lupus (deux cas), un syndrome de Sjögren (un cas), une polyarthrite rhumatoïde (un cas), un Lymphome amygdalien (un cas) et une tuberculeuse (un cas). Le motif d’hospitalisation était une pancytopénie fébrile dans 50 % des cas, une fièvre dans 20 % des cas, une pleurésie fébrile, un syndrome œdémateux, une cécité brutale dans respectivement 10 % des cas. Cliniquement, une altération de l’état général était trouvée chez sept patients, une fièvre chez 8, des frissons chez 3, une hépatomégalie chez 2, une splénomégalie chez 3, des adénopathies chez deux malades et une éruption cutanée chez quatre patients.
La numération de la formule sanguine a révélé une pancytopénie chez 80 % des patients : une anémie normochrome normocytaire chez 90 % des cas avec un taux moyen d’Hb à 7,6 g/dL, une leucopénie dans 80 % des cas, une thrombopénie chez tous les patients. D’autres anomalies biologiques étaient observées à type d’hypertriglycéridémie (60 %), un syndrome inflammatoire biologique (70 %), une cytolyse hépatique (40 %), une cholestase (50%) et des troubles de l’hémostase à type de CIVD chez un patiente. L’hyperferritinémie, vue chez huit patients, était à 3 840 en moyenne. Le myélogramme, de cellularité riche dans trois cas, normale dans trois cas, pauvre dans deux cas, a révélé de multiples images d’hémophagocytose dans 90 % des cas. Le SAM était secondaire à : un LES en poussée dans quatre cas, une maladie de Still (un cas), à une tuberculose pulmonaire et ganglionnaire (un cas), une mucormycose rhino-orbito-cérébrale (un cas), une toxicité au Méthotrexate (un cas) et était de cause indéterminée chez une patiente. Le traitement était aussi bien symptomatique qu’étiologique. On avait recours à une corticothérapie forte dose dans sept cas. L’évolution était bonne dans sept cas et fatale dans deux cas.
Conclusion. Cette étude montre que le syndrome d’activation macrophagique est une pathologie parfois insuffisamment évoquée dont l’issue peut être fatale. Les étiologies sont multiples pouvant être d’ordre infectieux, inflammatoire ou néoplasique. Les maladies systémiques sont relativement fréquentes dans notre série ceci pourrait être expliqué par un biais de recrutement. Les signes cliniques et biologiques restent peu spécifiques. C’est plutôt leur association qui doit amener le clinicien à évoquer ce syndrome précocement. Le traitement doit être démarré sans délai, parfois même avant d’avoir le diagnostic étiologique.
17-08
Thrombose veineuse profonde et anémie de Biermer
R. Ben Salah*, I. Chabchoub, M. Ben Hamad, F. Faten, M. Sameh, B. Zouhir
Médecine interne, CHU Hédi Chaker, Sfax, Tunisie
Introduction. La maladie veineuse thromboembolique (MVTE) est une pathologie fréquente mais également grave. Le traitement repose sur une anticoagulation efficace. Le défi pour le clinicien reste essentiellement la détermination de l’étiologie de cette MVTE puisque celle-ci conditionne la durée du traitement anticoagulant. L’anémie de Biermer constitue une cause rare de MVTE qu’il est important de la chercher.
Patients et méthodes. Nous avons colligé tous les cas de thrombose veineuse profonde révélant une maladie de Biermer colligées dans le service de médecine interne.
Résultats. Parmi 585 patients ayant une thrombose veineuse profonde (TVP), le diagnostic d’une anémie de Biermer a été retenu chez sept patients. Il s’agissait de trois hommes et quatre femmes. L’âge moyen était de 56 ans avec des extrêmes entre 44 et 76 ans.
La thrombose veineuse profonde était localisée aux membres inférieurs de siège proximal dans tous les cas étendus à la veine cave inférieur dans un cas. Elle était récidivante dans trois cas. Elle était compliquée d’une embolie pulmonaire dans cas.
La numération de la formule sanguine a montré une anémie macrocytaire dans six cas et une macrocytose isolée chez un patient. Une thrombopénie et une leucopénie associées ont été trouvées dans trois et un cas respectivement. Le dosage de l’homocystéinémie a été pratiqué dans six cas. Une hyperhomocystéinémie était retenue dans tous les cas. Elle était importante dans 60 % des cas.
Les anticorps anti-facteurs intrinsèques et anti-cellules pariétales, marqueur immunologique d’une anémie de Biermer étaient positifs dans deux cas.
Une atteinte neurologique type neuro-Biermer associée était retenue dans deux cas.
Sur le plan thérapeutique, tous les patients ont été traités par une anticoagulation curative associée à la vitamine B12. Cette vitaminothérapie a été prescrite par voie orale à la dose de 1 000 μg/jour. L’évolution était favorable dans tous les cas avec absence de récidive thromboembolique. Le traitement anticoagulant a pu être arrêté dans tous les cas après six mois avec absence de récidive de la MVTE.
Conclusion. La maladie de Biermer est une cause rare d’hyperhomocystéinémie acquise qui peut favoriser la survenue de thromboses veineuses. Il convient de l’évoquer chez tout patient présentant une thrombose veineuse sans cause évidente, principalement lorsqu’elle s’associe à une anémie macrocytaire.
17-09
Est ce que le groupe sanguin ABO est corrélé au risque de survenue d’hémopathie ?
F. Talbi*, S. Boudjalida, R. Abbadi, S. Menouer, F. Ardjoun, K. Djouadi
Hématologie, hôpital central de l’armée DR Mohamed Seghir Nekkache, Alger, Algérie
Introduction. De nombreuses études se sont intéressées à l’association entre le groupe sanguin et le risque d’hémopathies avec des résultats contradictoires ; nous avons évalué ce paramètre sur une série rétrospective (une année : juin 2015-2016). Les données sont recueillies sur un fichier Excel, l’analyse des variables est faite par le test du khi2 et la régression logistique.
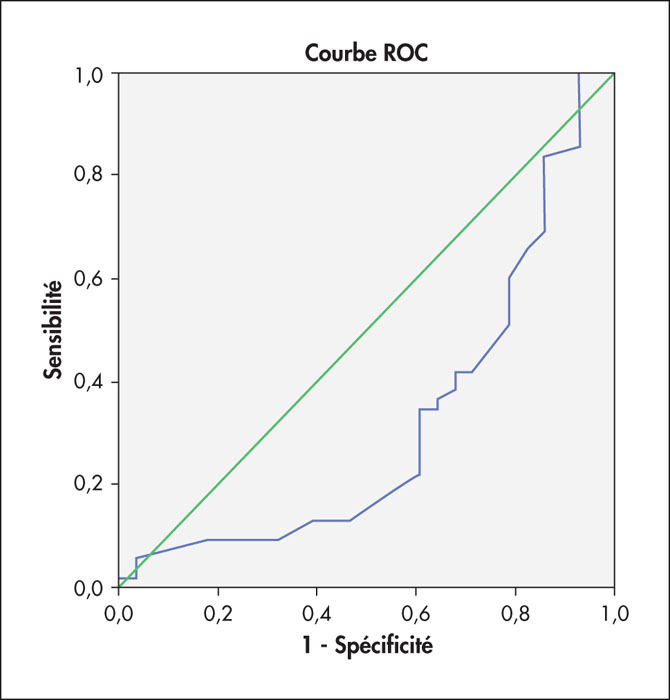
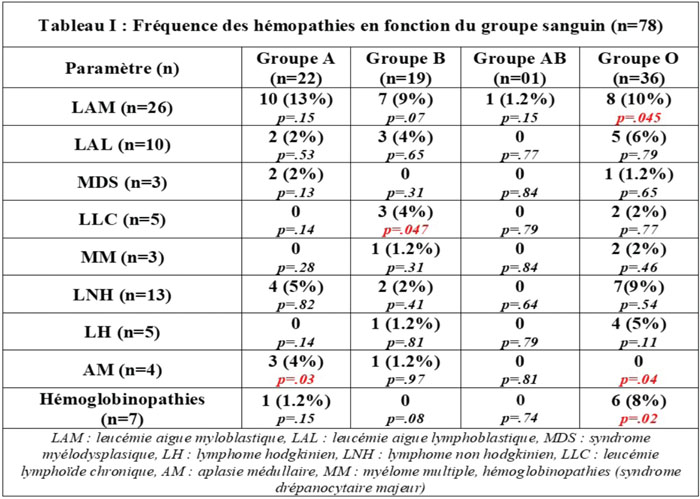
Résultats. N = 78 patients pris en charge dans l’unité clinique du service d’hématologie de l’hôpital central de l’armée : n = 50 H/28 F, sex-ratio : 1,78, l’âge moyen : 40 ± 16,3 ans (16-78). La corrélation entre l’âge, le sexe (tous types d’hémopathies confondus) → AUC : 0,38 [0,18-0,44], p = 0,007 (figure 1). La répartition des patients selon le type d’hémopathie et du groupe sanguin est illustrée dans le tableau 1.


Conclusion. Notre étude indique qu’il existe une corrélation entre groupe sanguin et le risque de survenue de certaines hémopathies : le seuil est significatif entre le groupe O et la LAM (p = 0,045), l’AM (p = 0,04) et la drépanocytose : p = 0,02) ; entre le groupe B et la LLC (p = 0,047) et entre le groupe A et l’AM (p = 0,03). Ainsi, malgré la taille réduite de notre échantillon, nous retrouvons des résultats qui rejoignent ceux de la littérature, ces données doivent être confirmées sur des séries exhaustives afin d’évaluer ce facteur (dans notre région). Par ailleurs, l’âge et le sexe sont des facteurs de risque connus en hématologie selon de nombreuses études épidémiologiques, cet élément est retrouvé dans notre cohorte.
17-10
Efficacité et tolérance de le romiplostim dans le traitement de purpura thrombopénique immunologique
N. Zatout*1, S. Hamdi2, H. Hamouda3
1 hématologie, CHU Saadna Abdenour de Sétif, Sétif, Algérie ; 2 Hématologie, CHU Saadna Abdenour Sétif, Sétif, Algérie ; 3 Hématologie, CHU Saadna Abdenour Sétif, Sétif, Algérie
Introduction. Le purpura thrombopénique immunologique (PTI) est une pathologie caractérisée par des mécanismes physiopathologiques multiples et complexes ; une destruction périphérique des plaquettes, un défaut de production médullaire prouvé actuellement par les effets cliniques des agonistes du TPO-R et enfin l’influence de facteurs génétiques et environnementaux.
Objectif : Évaluer l’efficacité et la tolérance du Nplatedans le PTI primaire chronique réfractaire ou nouvellement diagnostiqué.
Patients et méthodes. C’est une étude prospective incluant neuf patients (pts) : huit pts présentant un PTI réfractaire : La splénectomie a été réalisée en deuxième intention pour sept pts et en troisième intention pour un pt après échec au rituximab, une réponse transitoire a été notée dans un cas. Les traitements médicamenteux antérieurement reçus avec échec étaient le danazol (n : 1), le rituximab (n : 6), les immunosuppresseurs (n : cinq avec 2 réponses transitoires). Et une pte présente un PTI nouvellement diagnostiqué avec échec au traitement de première ligne et au rituximab. Le délai entre le diagnostic et le début de traitement par le Nplate est 10 ans (deux mois-20 ans), cinq hommes et quatre femmes avec un Sex-ratio (H/F) : 1,25, l’âge moyen à l’inclusion était de 31 ans (19-46). Le N-plate est administré à raison de un mg/kg/semaine en SC avec augmentation de la dose par palier hebdomadaire de 1 μg/kg jusqu’à un taux de plaquettes 350 × 109/L.
La réponse plaquettaire durable : Taux de plaquettes ≥ 50 × 109 pendant au moins six semaines de traitement, la Rce PLQ transitoire : Tde plaquettes ≥ 50 × 109 pendant au moins quatre semaines.
Résultats. À L’inclusion, le syndrome hémorragique cutané était rapporté chez quatre pts (44 %), les hémorragies des muqueuses chez deux pts (22 %), le score hémorragique était à quatre chez deux pts, 9 chez un pt, 15 chez un pt et nulle chez cinq pts. le taux moyen des plaquettes était 11 × 109/L (2-22). Tous les pts étaient sous corticoïdes à DME. Le délai moyen de l’obtention d’une réponse plaquettaire (≥ 50 × 109) était de six semaines (2-16), à la dose efficace moyenne de 4 μg/kg/j (2-9).
Une réponse plaquettaire durable est obtenue chez six pts (67 %) dont une pte nouvellement diagnostiquée, une réponse transitoire chez un pt (11 %), un échec chez 2 (22 %) pts, le taux de réponse globale est de78 %. Les traitements concomitants (corticoïdes, Ig polyvalents) ont pu être suspendus chez six pts. Les effets indésirables sont plus remarqués au début du traitement : asthénie et fatigue chez tous les pts, des douleurs osseuses ou arthralgies (n : 4), vertiges (n : 3), palpitations (n : 3), céphalées (n : 2), diarrhées (n : 2), fièvre (n : 2), épigastralgies (n : 1), nausées (n : 1), myalgies (n : 1), paresthésie (n : 1), thrombocytose (n : 3). On note des rechutes fréquentes à l’arrêt du traitement en cas de thrombocytose et même à la réduction des doses et des fluctuations de plaquettes sous une dose stable.
Conclusion. LeNplate est une approche thérapeutique efficace dans le PTI réfractaires et nouvellement diagnostiqué cependant suspensif et non curatif et coûteux. Les effets secondaires à court terme, le mode et le rythme d’administration sont gérables, cependant le risque de thrombose et de myélofibrose à long terme sont à surveiller.
17-11
Aspects épidémiologiques, cliniques, étiologiques et évolutifs de la thrombopénie en hématologie CHU Oran
R. Mrabet*1, R. Boukort2, D. Saidi3
1 HEMATOLOGIE, CHU ORAN, Oran, Algérie ; 2 Hématologie, CH et Universitaire d’Oran, Oran, Algérie ; 3 Hématologie, CH et Universitaire d’Oran, Oran, Algérie
Introduction. La thrombopénie constitue un motif de consultation fréquent en hématologie. Elle impose une démarche diagnostique spécifique et implique avant tout la vérification de sa véracité. Elle peut en fonction des facteurs de risque constituer une urgence thérapeutique. Parmi les étiologies, le PTAI occupe une place prépondérante.
Patients et méthodes. Étude rétrospective incluant des patients diagnostiques et suivis par le service d’hématologie du CHU Oran de janvier 2017 à décembre 2019.
Résultats. Sur 79 dossiers répertoriés, quatre patients présentaient une fausse thrombopénie et ont été exclus de l’étude. Parmi les 75 patients restants, 26 étaient des hommes et 49 des femmes soit un sr de 0,53. L’âge moyen au moment de diagnostic était de 39 ans avec des extrêmes variant de 16 ans à 79 ans. 20 % des patients provenaient du service de gynécologie. Les orientations étaient faites pour ménométrorragies ou grossesse évolutives. 14 % des patients étaient orientés par la cardiologie où ils étaient majoritairement suivis pour HTA. Les autres orientations ont été faites par des médecins généralistes, infectiologues, chirurgiens et internistes. Dans 68 cas, il s’agissait d’une thrombopénie nouvellement diagnostiquée et dans huit cas il s’agissait d’une thrombopénie persistante. Un syndrome hémorragique était présent au moment du diagnostic dans 65 % des cas et le score de Khelaf était en moyenne de 8 avec des extrêmes variant de trois à 13. Le taux moyen de plaquettes était de 68000/mm3 et le volume moyen des plaquettes était de 12. Le médullogramme a confirmé l’origine périphérique chez tous les patients et l’absence d’envahissement. Le bilan d’hémostase pratiqué chez tous les patients était correct.
Une cause iatrogèene a été retenue chez cinq patients. 2 causes infectieuses ont été retenues : une liée à une infection CMV et la seconde à un HIV. Quatre cas de maladie de systèeme ont été retenues après bilan immunologique concluant.
Une abstention thérapeutique avec surveillance raprochée a été indiquée chez 35 % des patients. Le traitement a comporté des immunoglobulines et une corticothéerapie chez cinq patients, une corticothéerapie seule chez 21 patients.
Une corticorésistance a été constatée chez huit patients suivie d’un échec au rituximab. Une rémission a été obtenue aprèes spléenectomie.
Conclusion. Le PTAI atteint préférentiellement les femmes jeunes. Il s’agit d’un diagnostic d’élimination, aussi la démarche se doit d’êetre méthodique et la plus complèete possible.
17-12
Groupes sanguins dans le système ABO et le diabète de type 2
A. Sall*1, S. Diao2, M. Seck1, BF. Faye1, FB. Sall1, S. Fall1, M. Gadji1, FS. Ndiaye1, S. Diop1, FAO. Touré1
1 Hématologie, Université Cheikh Anta Diop, Dakar, Sénégal ; 2 Hématologie, CHU Aristide Le Dantec, Dakar, Sénégal
Introduction. Maladie métabolique, le diabète de type 2 constitue un problème de santé publique. Selon la Fédération internationale de diabète (IDF), il y aurait 427 millions de diabétiques dans le monde soit une prévalence de 8,5 %. La prédisposition génétique, l’obésité, la sédentarité jouent un rôle prépondérant dans l’apparition de la maladie mais un autre facteur de risque a été identifié : le groupe sanguin ABO. L’objectif général de notre étude était de rechercher un lien entre les groupes sanguins ABO et le diabète de type 2.
Matériels et méthodes. Nous avons effectué une étude cas-témoins sur une période de deux ans. Les cas, diabétiques, étaient suivis dans différents centres de diabétologie de Dakar. Les témoins, indemnes de diabète, ont été recrutés parmi les donneurs de sang du Centre national de transfusion sanguine (CNTS) de Dakar. Un prélèvement sur tube avec anticoagulant a été effectué chez les diabétiques afin de déterminer le groupe sanguin ABO alors que deux tubes (EDTA et fluorure de sodium) ont été nécessaires chez les témoins non diabétiques pour déterminer respectivement le groupe sanguin ABO et la glycémie à jeun. Les groupages sanguins ont été effectués sur carte gel (Bio-Rad, France) et la glycémie até déterminée sur l’automate Architect (Abbott, USA). Tous les participants ont signé un consentement libre et éclairé avant d’être enrôlés dans l’étude.
Résultats. Au terme de deux ans, 313 diabétiques et 313 témoins ont été inclus dans notre étude. L’âge moyen des diabétiques était de 54,5 ans [20-87 ans] avec une nette prédominance féminine (sex-ratio = 0,4).
La reparution des groupes sanguins ABO dans notre population d’étude était :
- –diabétiques : groupe O = 47%, groupe AB = 12%, groupe A = 20%, groupe B = 21%.
- –témoins : groupe O = 56,9%, groupe AB = 4,1%, groupe A = 23%, groupe B = 16%.
On notait une différence significative entre les groupes AB (p = 0,01) et OO (p = 0,013) avec des OR respectifs de 2,99 [1,56-5,77] et 0,66 [0,48-0,91]. Pour les groupes A et B dans les deux populations, les calculs ne révélaient pas de lien significatif.
Conclusion. Notre étude révèle que le groupe sanguin AB multiplie par 3 le risque de survenue de diabète de type deux alors que le groupe O est protecteur.
D’autres études avec un échantillonage plus important devront être menées pour conforter ces résultats mais également pour déterminer par quel mécanisme les groupes sanguins ABO favorisent ou non la survenue du diabète de type 2.
17-13
Évaluation de la prévalence de l’allo-immunisation fœto-maternelle anti-RH1 au CHU de Blida (Algérie)
N. Haddad*1, Y. Guedjal2, A. Bailiche2, A. Belkheiri2, Y. Sadi3, F. Saadaoui3, L. Kedji3, S. Abdi1
1 Laboratoire central, CHU Blida, Faculté de Médecine, Université Blida1, Blida, Algérie ; 2 Département de pharmacie, Faculté de médecine, université Blida1, Blida, Algérie ; 3 Pédiatrie, CHU Blida, Faculté de médecine, Université Blida1, Blida, Algérie
Introduction. L’allo-immunisation fœto-maternelle (AIFM) anti-RH1 est la première cause d’anémie fœtale néonatale. Elle est responsable d’un syndrome hémolytique fœtal de gravité variable, pouvant se manifester par un ictère hémolytique néonatal, une anasarque fœtoplacentaire, voire une mort in utero.
De nombreux progrès ces dernières années ont permis de diminuer son incidence. Ces progrès ont porté essentiellement sur la généralisation de l’injection prophylactique d’immunoglobuline anti-RH1 dans les 72 h qui suivaient l’accouchement. Depuis peu, cette prophylaxie a été renforcée par une injection supplémentaire à la 28e semaine de gestation pour prévenir les éventuelles allo-immunisations au cours de la grossesse.
L’objectif de l’étude est d’évaluer la prévalence de l’AIFM anti-RH1 dans la région de Blida.
Patients et méthodes. Il s’agit d’une étude transversale descriptive sur une période de trois ans allant de janvier 2016 à décembre 2018.
Toutes les femmes RH-1 adressées au laboratoire d’hémobiologie du CHU de Blida à l’unité Hassiba Benbouali pour la recherche d’agglutinines irrégulières (RAI) ont été incluses.
La technique utilisée est celle du Coombs indirect sur tube.
Les RAI positives pour l’anti-RH1 au dépistage, ont été suivies par une étape de titrage.
Résultats. Sur 538 RAI réalisées chez ces femmes RH-1 durant la période d’étude, 18 étaient positives pour l’anti-RH1, soit une prévalence de 3,34 %.
L’âge moyen des femmes immunisées est de 32,5 ans (22-41 ans).
61 % des cas sont diagnostiquées devant un test de Coombs direct positif chez le nouveau-né RH+1 avec un ictère précoce.
Le titre de l’anti-RH1 était > 1/16 (seuil du risque d’atteinte fœtale) dans 11 cas (un cas : 1/2048, trois cas : 1/256, deux cas : 1/128, deux cas : 1/64, trois cas : 1/32).
L’enquête étiologique de cette allo immunisation a révélé que 44,4 % des femmes n’ont pas reçu la prophylaxie anti-RH1 au moment des accouchements ou des avortements antérieurs.
Concernant les antécédents obstétricaux de ces femmes, on retrouve : mort in utero (02 cas), morts-né dans un tableau d’anasarque fœtoplacentaire (deux cas), enfant avec ictère néonatal nécessitant une exsanguinotransfusion (cinq cas), un enfant avec un ictère néonatal compliqué d’une infirmité motrice cérébrale (un cas) et un cas d’avortement à répétition.
Conclusion. L’AIFM anti-RH1 peut mettre en péril l’avenir obstétrical des femmes RH-1. Sa prévalence reste non négligeable à Blida. Le dépistage systématique des femmes à risque selon le calendrier des RAI durant la grossesse, et la prophylaxie anti-RH1 de la 28e semaine peut réduire considérablement son incidence.
17-14
Transfusion de plaquettes Rhésus incompatibles en milieu d’oncohématologie : à propos de 48 patients
S. Fekih Salem*, EB. Wejden, F. Ben Lakhal, E. Gouider
Laboratoire d’hématologie biologique, Hôpital Aziza Othmana, Tunis, Tunisie
Introduction. Les antigènes du système Rhésus ne sont pas exprimés sur les plaquettes, néanmoins une allo-immunisation antiérythrocytaire peut être induite après transfusion de concentrés plaquettaires (CP), du fait de la présence d’hématies résiduelles. Le but de l’étude était d’analyser les caractéristiques des CP transfusés aux patients atteints de leucémies aiguës (LA) et d’évaluer la proportion d’allo-immunisations antiérythrocytaire anti-Rhésus.
Patients et méthodes. Il s’agit d’une étude prospective (octobre 2018/avril 2019). Tous les patients traités pour LA au service d’hématologie clinique ont été inclus. Les patients transférés ou décédés avant la réalisation du bilan immuno-hématologique ont été exclus. Pour chaque patient, nous avons relevé les données suivantes : âge, sexe, type de LA, groupe sanguin (GS) ABO, phénotype érythrocytaire Rhésus (RH1, RH2, RH3, RH4 et RH5). Des renseignements concernant la transfusion de plaquettes au cours de l’hospitalisation des patients ont été rapportés : quantité et type de CP (concentrés de plaquettes standards (CPS) ou concentrés de plaquettes d’aphérèse (CPA), groupage ABO et phénotype Rhésus. Nous avons réalisé pour chaque patient une recherche d’anticorps antiérythrocytaires (RAE) par technique de Coombs indirect en basse force ionique en : prétransfusionnel, à J15, à J30 d’hospitalisation et lors de consultations ultérieurs.
Résultats. 48 patients ont été inclus. L’âge moyen était de 38 ans, le sex-ratio était de 1,5, 72,9 % avaient une LAM. 56 % étaient de groupe sanguin O. Quatre patients étaient RH-1,2039 CP ont été transfusés durant l’étude : 99,2 % étaient des CPS. 45 % des CP transfusés étaient en incompatibilité ABO majeure. 94 % des CP transfusés aux patients RH-1 étaient issus de donneurs RH1, soit 37,75 CP/patient. 85,5 % des malades ont reçu des CP incompatibles dans les autres antigènes du système Rhésus. Aucun patient n’a développé d’alloanticorps après un suivi moyen de quatre semaines (1-40).
Conclusion. L’absence d’allo-immunisation antiérythrocytaire chez les patients immunodéprimés et notamment en oncohématologie a été rapportée dans la littérature. Néanmoins plusieurs facteurs la modulent (quantité d’hématies résiduelles, méthode de préparation des CP, période de suivi post-transfusionnelle). La transfusion de CP Rhésus compatible est difficile vu la difficulté d’approvisionnement et le délai de péremption mais l’immunoprophylaxie anti-RH1 chez les sujets de sexe féminin est recommandée ainsi qu’un suivi régulier par la RAE.
17-15
Caractéristiques épidémiocliniques et résultats thérapeutiques des anémies hémolytiques auto-immunes : expérience monocentrique
S. Kefi*, R. Jaïdi, Y. Khayati, A. Selmi, D. Jabr, R. Mansouri, H. Ben Neji, E. Berred, M. Bchir, M. Achour, Y. Ben Abdennebi, M. Bahri, R. Kharrat, K. Kacem, R. Ben Lakhal, B. Meddeb, L. Aissaoui
Hématologie clinique, Hôpital Aziza Othmana, Tunis, Tunisie
Introduction. L’anémie hémolytique auto-immune (AHAI) est une pathologie rare mais potentiellement grave liée à la présence d’autoanticorps dirigés contre un ou plusieurs antigènes érythrocytaires. La démarche diagnostique et thérapeutique dépend des propriétés immunochimiques des autoanticorps et de la présence ou non d’une maladie sous-jacente. L’objectif de notre travail était de rapporter les caractéristiques épidémiologiques et cliniques ainsi que les résultats thérapeutiques des patients traités pour une AHAI dans notre service.
Patients et méthodes. Nous avons mené une étude rétrospective et descriptive sur cinq ans de janvier 2013 jusqu’à décembre 2017. Tous les patients pris en charge pour une AHAI durant cette période ont été inclus. Nous n’avons pas inclus dans notre étude les syndromes d’Evans et les AHAI compliquant des syndromes lymphoprolifératifs déjà connus.
Résultats. Notre série a porté sur 67 patients. L’âge médian était de 53 ans [18-50] avec une légère prédominance féminine (sex-ratio à 0,56). La présentation clinique était hétérogène. Un ictère cutanéomuqueux, des signes de mal tolérance cardiaque, une hémoglobinurie et une splénomégalie ont été notés dans respectivement 33 %, 33 %, 28 % et 20 % des cas. Les taux médians respectifs d’hémoglobine, de réticulocytes, de Bilirubine totale et de lacticodéshydrogénase étaient de 5,9 g/dL, 247 000/mm3, 48 mmol/L et 481 UI/L. Le TCD était positif chez 97 % des patients. Une AHAI à autoanticorps chauds a été observée chez 71 % des patients et une AHAI à autoanticorps froids chez 21 % des patients. Une étiologie sous-jacente a été retrouvée chez 18 % des patients. Les étiologies étaient dominées par un lymphome indolent chez deux patients, un Lupus érythémateux disséminé chez deux patients et une pneumopathie à mycoplasme chez deux patients qui avaient une AHAI froide. Le traitement de première ligne était une corticothérapie chez tous les patients (Prednisolone 1 mg/kg/jour chez 80 % des malades et dexaméthasone chez 20 %). La totalité des patients ont reçu un traitement de support par de l’acide folique et 27 % ont eu une transfusion de concentrés de globules rouges phénotypés devant des signes de souffrance cardiaque. Une réponse complète après le traitement de première ligne a été obtenue chez 78 % de la population étudiée, une réponse partielle chez 11 % et un échec chez 11 %. L’âge > 50 ans et les autoanticorps chauds étaient des facteurs prédictifs de bonne réponse. Parmi les patients répondeurs, le taux de rechute était de 21 %.
Conclusion. L’AHAI reste une pathologie grave nécessitant une prise en charge urgente. La réalisation d’un bilan étiologique exhaustif et systématique à la recherche d’une maladie sous-jacente qui sera traitée le cas échéant, améliorera significativement la prise en charge thérapeutique.
17-16
Anémie hémolytique auto-immune : expérience du service d’hématologie clinique du CHU de Yopougon en côte d’Ivoire à propos de 30 cas
K. Ismael*1, K. Boidy1, N. Méité2, D. Silué3, J. Atcha3, RP. Botti3, A. Tolo1
1 Hématologie, Centre hospitalo-universitaire de Yopougon, Abidjan, Côte d’Ivoire ; 2 Hématologie clinique CHU Yopougon, CHU Yopougon, Abidjan, Côte d’Ivoire ; 3 Hématologie, CHU Yopougon, Abidjan, Côte d’Ivoire
Introduction. Les anémies hémolytiques auto-immunes (AHAI) sont les plus fréquentes des anémies hémolytiques acquises mais restent une cause rare de maladie auto immune. Notre objectif est de contribuer à l’étude des AHAI.
Patients et méthodes. Il s’est agi d’une étude rétrospective et descriptive sur une période de 30 ans, qui s’est déroulé au service d’hématologie clinique du CHU de Yopougon. Les patients présentant une anémie hémolytique avec un test de Coombs direct positif et un dossier médical complet ont été inclus. Les données sociodémographiques, cliniques, biologies, étiologiques et thérapeutiques ont été recueillies.
Résultats. 30 patients ont été inclus. L’âge moyen était de 39 ans avec des extrêmes de 7 à 78 ans. Le sex-ratio était de 0,42. La pâleur était retrouvée dans 83,3 % des cas, l’ictère dans 53,3 % des cas. Le taux moyen d’hémoglobine était de 5,8 g/dL [3,9-8,1]. Le taux moyen de bilirubine total était de 18,3 mg/L [1,5-48], celui de la bilirubine libre était de 11,69 mg/L [0,9-30] et celui des LDH 857 UI/L [208-1780]. L’étiologie était idiopathique chez 19 patients (63,34 %), sept cas de syndrome lymphoprolifératifs chronique (20 % LLC et 3,33 % Lymphome de malt), trois cas d’hépatite virale B (10 %) et un cas de LED (3,33 %). Les traitements utilisés étaient la corticothérapie (100 %), les immunosuppresseurs (3,3 %), et le Mabthéra (20 %). 13,33 % de décès étaient observés (anémie 25 %, LLC 25 %, inconnue 50 %).
Conclusion. L’AHAI est relativement rares. L’étiologie est dominée par les causes idiopathiques dans notre contexte d’exercice. Le Mabthéra a amélioré l’évolution des formes dites réfractaires.
17-17
Recherche des agglutinines irrégulières en milieu obstétrical : à propos de 452 cas
S. Bouzidi*1, A. Nasr1, F. Jarraya1, R. Arfaoui2, S. Mahfoudh1, M. Bouguerra1, S. Layouni1, N. Fekih-Mrissa3, R. Rachdi2, B. Nsiri1
1 Laboratoire d’hématologie, Hôpital militaire de Tunis, Tunis, Tunisie ; 2 Service de gynécologie-obstétrique, Hôpital militaire de Tunis, Tunis, Tunisie ; 3 Unité de recherche de biologie moléculaire (ur17dn06), laboratoire d’hématologie, Hôpital militaire de Tunis, Tunis, Tunisie
Introduction. L’incompatibilité fœto-maternelle érythrocytaire reste la première cause d’anémie fœtale et néonatale. D’où l’importance de la surveillance de toute grossesse et du dépistage de l’allo-immunisation chez la femme enceinte afin d’intervenir au bon moment.
Matériels et méthodes. Il s’agit d’une étude rétrospective portant sur la recherche d’agglutinines irrégulières (RAI) chez des femmes enceintes suivies au service de gynécologie obstétrique dans l’hôpital militaire principal d’instructions de Tunis, sur une période de trois ans (janvier 2016-décembre 2018).
Pour chaque femme enceinte ayant une RAI positive, nous avons recueilli l’identité des patientes, l’âge, les antécédents, le nombre de grossesses, la parité, et le terme de la grossesse actuelle. Nous avons aussi noté le groupe sanguin ABO, le phénotype érythrocytaire Rhésus-Kell, et la notion d’injection d’immunoglobulines anti-Rh1.
Le panel utilisé pour la réalisation de la RAI (dépistage et identification) est officiellement défini (circulaire 49/05). La technique utilisée est le test de Coombs indirect à basse force ionique sur carte gel LISS/Coombs et la technique enzymatique.
Résultats. Au total, 452 femmes enceintes ont bénéficié de 495 RAI. Cette dernière a été positive chez 21 femmes (4,6 %). L’âge moyen était de 31 ans (22-37 ans). Le groupe sanguin O était le plus fréquent (47,6 %), ainsi que le phénotype Rh1 négatif (81 %). Treize alloanticorps identifiés étaient de type anti-Rh1, soit 61,9 %. Une association d’allo-anticorps a été observée dans deux cas (9,5 %). L’origine de l’allo-immunisation érythrocytaire était prédominée par les avortements dans sept cas (33,3 %), et les métrorragies dans quatre cas (19 %). Par ailleurs, trois femmes (14,3 %) n’avaient aucun antécédent transfusionnel ni obstétrical. L’immunoprophylaxie anti-Rh1 a été mentionnée chez sept femmes (33,3 %).
Conclusion. L’immunisation anti-Rh1 est la plus fréquente dans la population étudiée. Cela pourrait résulter d’une insuffisance du suivi des grossesses et/ou l’utilisation de protocoles d’immunoprophylaxie mal adaptés.
17-18
Syndrome d’activation macrophagique de l’adulte en milieu de réanimation : à propos de 30 cas
S. Bouzidi*1, A. Rafrafi2, W. Sallemi2, A. Nasr1, S. Layouni1, N. Fekih-Mrissa3, M. Ferjani2, B. Nsiri1
1 Laboratoire d’hématologie, Hôpital militaire de Tunis, Tunis, Tunisie ; 2 Service d’anesthésie réanimation, Hôpital militaire de Tunis, Tunis, Tunisie ; 3 Unité de recherche de biologie moléculaire (ur17dn06), laboratoire d’hématologie, Hôpital militaire de Tunis, Tunis, Tunisie
Introduction. Le syndrome d’activation macrophagique (SAM) est une maladie rare potentiellement mortelle, lié à une stimulation inappropriée des cellules macrophagiques dans la moelle osseuse et le système lymphoïde, ce qui entraîne une phagocytose anormale des éléments figurés du sang et la libération de cytokines pro-inflammatoires. Le but de notre étude est de décrire les particularités épidémiologiques, diagnostiques et thérapeutiques du SAM dans un service de réanimation polyvalente.
Matériels et méthodes. Il s’agit d’une étude rétrospective, descriptive sur une période de cinq ans, colligeant 30 patients hospitalisés dans le service de réanimation de l’Hôpital Militaire de Tunis et diagnostiqués avec SAM. Tous les patients inclus avaient un H-score supérieur à 169 et une image d’hémophagocytose au myélogramme.
Résultats. L’âge moyen des patients était de 49 ans ± 17 ans avec une prédominance féminine. Cinq patients étaient immunodéprimés (sous traitement corticoïde). Les motifs d’admission les plus fréquents étaient les états de choc et les détresses neurologiques. Le signe clinique le plus fréquent était la fièvre (18 cas). L’anomalie biologique la plus fréquente sur l’hémogramme était la bicytopénie présente chez 17 patients (anémie et thrombopénie). Le H-score moyen était égal à 209. Les défaillances hémodynamiques et respiratoires étaient les plus fréquentes. Le traitement par corticoïdes était entrepris chez 14 patients alors que 13 patients ont bénéficié des immunoglobulines. Un seul patient a été mis sous étoposide. L’infection était l’étiologie la plus fréquente du SAM avec prédominance des infections bactériennes à bacilles multirésistants. La mortalité à J28 d’hospitalisation était de 40 %. La cause de décès la plus fréquente était le choc septique. Les scores SOFA et APACHE II étaient les seuls facteurs prédictifs de mortalité (p = 0,015 et p = 0,042).
Discussion. Ce syndrome peut être primaire essentiellement chez l’enfant, ou secondaire à diverses affections, hématologiques, infectieuses ainsi qu’à des maladies auto-immunes variées. Le diagnostic repose sur l’association de signes cliniques et biologiques, non spécifiques. La mise en évidence de l’hémophagocytose à la cytologie ou à l’histologie permet de le confirmer. Le H-score pourrait être d’un grand apport dans l’identification précoce des patients ayant un SAM en réanimation et ainsi une prise en charge rapide.
Conclusion. Le SAM reste un syndrome grave. Le pronostic est sombre, avec une mortalité de près de 50 % toutes causes confondues. Il doit être évoqué devant tout tableau fébrile associé à une bi- ou une pancytopénie. Le traitement est encore mal codifié et doit être démarré sans délai, parfois même avant d’avoir le diagnostic étiologique. La précocité du diagnostic conditionne le pronostic et impose une prise en charge multidisciplinaire.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License