Hématologie
MENU10 Néoplasies myéloprolifératives Ph- Volume 26, supplement 4, Septembre 2020

10-01
Caractéristiques cliniques, évolution à long terme et facteurs prédictifs de rechute après arrêt de l’Imatinib au cours de leucémie chronique à éosinophiles associée au réarrangement FIP1L1-PDGFRA : étude rétrospective à propos de 151 patients
J. Rohmer*1, A. Couteau-Chardon2, JM. Cayela3, L. Mauvieux4, MJ. Mozziconacci5, E. Delabesse6, P. Flandrin-Gresta7, C. Godon8, A. Bidet9, R. Ben Abdelali10, M. Hunault-Berger11, L. Galicier12, M. Hamidou13, E. Lengline14, C. Mohr15, FE. Nicolini16, J. Rey17, L. Terriou18, S. Tavitian19, I. Machelart20, B. Arnulf21, A. Guffroy22, F. Lhomme23, N. Grardel24, P. Cony-Makhoul25, G. Lefèvre26, C. Preudhomme27, JE. Kahn28, M. Groh29
1 Médecine Interne Cereo, Hôpital Foch, Suresnes ; 2 Centre de référence des syndromes hyperéosinophiliques, médecine interne, Hôpital Foch, Suresnes ; 3 Laboratoire d’hématologie, Hôpital Saint-Louis (AP-HP), Paris ; 4 Laboratoire hématologie, CHU de Strasbourg, Strasbourg ; 5 Laboratoire de biopathologie, Institut Paoli-Calmettes, Marseille ; 6 Oncopole, CHU Toulouse - Casselardit Ancely, Toulouse ; 7 Laboratoire hématologie, CHU de Saint-Étienne, Saint-Priest-en-Jarez ; 8 Laboratoire hématologie biologique, Hôtel-Dieu, Nantes ; 9 Laboratoire d’hématologie, CHU de Bordeaux, Bordeaux ; 10 Oncohématologie, Laboratoire Cerba, Saint Ouen l’Aumône ; 11 Service des maladies du sang, CHU - CHU Angers, Angers ; 12 Immunologie clinique, CHU Saint-Louis, Paris ; 13 Médecine interne, CHU de Nantes, Nantes ; 14 Département d’hématologie, Hôpital Saint-Louis, Paris ; 15 Hématologie clinique, CHU La Réunion, Saint-Pierre ; 16 Hématologie, Centre Léon Bérard, Lyon ; 17 Hématologie, Institut Paoli-Calmettes, Marseille ; 18 Unité d’immunologie clinique, EA2686, CHRU Lille, Lille ; 19 Hématologie, Institut Universitaire du Cancer Toulouse Oncopole, Toulouse ; 20 Médecine interne, médecine interne, Bordeaux ; 21 Hématologie, Assistance Publique Hôpitaux de Paris, Paris ; 22 Department of Clinical immunology and Internal Medicine, National Reference Center for Autoimmune Diseases, Hôpitaux Universitaires de Strasbourg, Strasbourg ; 23 Hématologie, CHU de Rennes, Rennes ; 24 Laboratoire d’hématologie, CH Régional Universitaire de Lille, Lille ; 25 Centre de recherche clinique, CH Annecy Genevois, Metz-Tessy ; 26 Institut d’immunologie, réseau éosinophile, EA2686, CHRU Lille, Lille ; 27 Centre de biologie pathologie - laboratoire hématologie, CH Régional Universitaire de Lille, Lille ; 28 Médecine interne, Cereo, Suresnes ; 29 Cereo (centre de référence des syndromes hyperéosinophiliques) - médecine interne, Hôpital Foch, Suresnes
Introduction. L’Imatinib mésylate (IM) est le traitement de choix au cours de la leucémie chronique à éosinophiles associée au transcrit FIP1L1-PDGFRA (LCE F/P+) avec une possibilité d’arrêt de traitement sans qu’aucun facteur de rechute n’ait été mis en évidence.
Patients et méthodes. Étude française multicentrique rétrospective incluant les patients avec une LCE F/P+ entre 2003 et 2016. Les objectifs étaient de décrire les caractéristiques clinicobiologiques ainsi que l’évolution des patients à long terme en «vie réelle» de ces patients, fournir des données d’incidence à une échelle nationale et d’identifier des facteurs prédictifs de rechute après arrêt de l’IM à l’aide d’un modèle de régression logistique.
Résultats. Parmi les 151 patients inclus, 143 (95 %) étaient des hommes (âge moyen 49 ans). L’incidence annuelle de la maladie était de 0,18 cas par an par million d’habitants. Les organes les plus fréquemment atteints étaient la rate (45 %), la peau (32 %), le poumon (30 %), le cœur (19 %) et le SNC (9 %). Le pic d’éosinophilie moyen était de 10,3 (± 6) G/L. Une anémie et une thrombopénie étaient observées dans 34 (24 %) et 42 (28 %) des cas respectivement tandis que 47 (31 %) patients avaient une numération normale en dehors de l’éosinophilie. Les dosages de vitamine B12 et de tryptase sérique étaient élevés dans 74/79 (94 %) et 45/57 (79 %) des cas tandis que le dosage de CRP était > 40 mg/L chez cinq (4 %) patients. La corticothérapie n’a permis d’obtenir une rémission hématologique complète (RHC) chez aucun patient. Tous les patients traités par imatinib ont eu une réponse hématologique (n = 146) et moléculaire (n = 84 tests) complète. La durée de suivi moyenne était de 80 (±56) mois avec une survie globale à 1, cinq et 10 ans de 99 %, 95 % et 84 % respectivement. Parmi les 46 patients ayant arrêté l’IM, 20 ont présenté une rechute permettant de définir un taux de rémission sans traitement de 57 %. En analyse multivariée, un amaigrissement (HR : 5,98 95 % CI [1,63-21,89] ; p = 0,004), le délai d’introduction du traitement (HR 1,02 [1,00-1,03] ; p = 0,01) et une courte durée de traitement (HR : 0,97 [0,95-0,99] ; p = 0,01) était des facteurs associés à la rechute. Tous les patients qui ont repris un traitement par IM après une rechute ont de nouveau présenté une réponse complète.
Conclusion. Cette étude avec un large effectif confirme la prédominance masculine de cette pathologie ainsi que la fréquence des atteintes respiratoire, cardiaque, cutanée et splénique. Elle confirme l’excellente efficacité de l’IM et le pronostic favorable à long terme. Après arrêt de l’IM, un amaigrissement, une courte durée de traitement préalable et un délai prolongé d’introduction de l’IM étaient associés à la rechute.
10-02
BCL-2 est exprimée dans les mastocytoses systémiques avancées et la midostaurine induit une sensibilité au vénétoclax
J. Moraly*1, D. Canioni2, L. Lhermitte3, L. Polivka4, J. Rossignol5, M. Feroul6, S. Fraitag7, H. Bouktit8, L. Frenzel9, M. Arock10, P. Dubreuil11, L. Maouche-Chrétien12, O. Hermine5
1 Hématologie clinique, Hôpital Necker, Paris ; 2 Service de cytologie et anatomie pathologiques, Hôpital Necker, Paris ; 3 Laboratoire hématologie biologique, hôpital Necker, Paris ; 4 Laboratoire Pr Hermine, Institut Imagine, Paris ; 5 Hématologie, hôpital Necker, Paris ; 6 Laboratoire Inserm U1151, Inem, Paris ; 7 Laboratoire de pathologie, Hôpital Necker, Paris ; 8 Ceremast (centre de référence des mastocytoses), Hôpital Necker, Paris ; 9 Hématologie clinique, AP-HP, Paris ; 10 Pharmacie, École normale supérieure de Cachan, Paris ; 11 CRCM, U1068 Inserm, Marseille ; 12 Laboratoire Inserm U1153, Institut Imagine, Paris
Introduction. Les mastocytoses systémiques (MS) sont un groupe hétérogène de pathologies rares caractérisées par une accumulation de mastocytes dans un ou plusieurs organes, et associées à des mutations du gène KIT (KIT D816V) dans plus de 90 % des cas. Les MS comprennent des formes indolentes (MSI) et des formes avancées (MSA). Dans les MSA, la Midostaurine permet d’obtenir un taux de réponse élevé et une amélioration du pronostic. Néanmoins, les réponses thérapeutiques sont rarement complètes et durables. Des combinaisons thérapeutiques sont donc à envisager afin d’optimiser ces résultats. Le vénétoclax est un inhibiteur sélectif de BCL-2, capable de lever la résistance à l’apoptose induite par BCL-2 et d’induire la mort cellulaire. Notre objectif était d’évaluer l’expression de BCL-2 dans les MS indolentes et avancées, d’évaluer la dépendance aux protéines BH-3 in vitro ainsi que la sensibilité au vénétoclax seul ou en association avec la Midostaurine.
Matériels et méthodes. Trente-trois patients ont été inclus dans cette étude préliminaire. Les patients ont été classés selon la classification OMS en MS indolente (MSI, n = 10), MS smoldering (MSS, n = 1) et MS avancées (MSA, n = 20), parmi lesquelles neuf MS associées à une hémopathie (MS-AHN), quatre leucémies à mastocytes (MCL), et deux sarcomes mastocytaires (SMC). La majorité des patients avaient une mutation de KIT D816V (n = 30 ; 90,9%), deux MCL et un MCS avaient respectivement une mutation du domaine extracellulaire et juxtamembranaire de KIT. Neuf patients ont été traités avec la Midostaurine en première ligne. Des biopsies ostéomédullaires ont été réalisées au diagnostic et pendant le suivi. Les mastocytes étaient identifiés à l’aide des marqueurs CD117 et tryptase en IHC. Le marquage BCL-2 était annoté négatif ou positif (> 5% des cellules) ; rare, hétérogène ou homogène (> 80% des cellules positives) ; d’intensité faible ou forte (≥ ou < aux lymphocytes T réactionnels). Un BH3 profiling sur les lignées ROSA KIT WT et ROSA KIT D816V, après exposition à différents BH3-inhibiteurs et après exposition à la Midostaurine a été réalisé.
Résultats. Dans les MSI, BCL-2 était négatif (n = 2/10) ou faiblement exprimé (n = 8/10). À l’inverse, dans les MSA, BCL-2 était constamment exprimé (16/16) avec une intensité forte (13/16) et parfois homogène (6/16). Dans les MCL et SMC, BCL-2 était toujours positif homogène et fortement exprimé. Chez les patients traités par Midostaurine, le marquage BCL-2 réalisé avant traitement et après trois mois était resté inchangé, en dépit de la diminution de l’infiltration mastocytaire. In vitro dans les lignées mastocytaires MCL-like ROSA KIT WT et ROSA KIT D816V, l’expression des protéines BCL-2, MCL-1 et BCL-XL a été évaluée à l’état basal puis après traitement par la Midostaurine (200 nM, 48 h). Sous Midostaurine, l’expression de BCL-XL et de MCL-1 était significativement réduite. De façon cohérente avec les données in vivo sur les patients, l’expression de BCL-2 sous Midostaurine était restée inchangée. De plus, les données de BH-3 profiling après traitement par la Midostaurine ont pu mettre en évidence une augmentation de la dépendance apoptotique à BCL-2, particulièrement dans la lignée ROSA KIT D816V.
Conclusion. BCL-2 est fortement exprimée dans les mastocytoses systémiques avancées et pourrait participer à la pathogenèse de la maladie, à sa chimiorésistance et à sa résistance partielle à la Midostaurine. Nos résultats fournissent un rationnel à l’utilisation de la Midostaurine et du vénétoclax en combinaison dans les mastocytoses avancées.
10-03
Prédiction de l’efficacité à long terme de l’interféron α dans les néoplasmes myéloprolifératifs mutés JAK2V617F et CALR
A. Tisserand*1, R. Noble2, M. Mosca1, C. Marzac3, G. Vertenoeil4, H. Compario5, M. El Khoury1, C. Marty1, A. Di Stefano1, N. Casadevall6, E. Solary1, F. Pasquier1, H. Raslova1, B. Cassinat7, S. Constantinescu4, JJ. Kiladjian8, F. Girodon5, M. Hochberg2, JL. Villeval1, W. Vainchenker1, I. Plo1
1 Inserm, UMR1287, Université Paris-Sud, Gustave Roussy, Villejuif ; 2 Institut des sciences de l’évolution, Université de Montpellier, Montpellier ; 3 Hématologie biologique, Gustave Roussy, Villejuif ; 4 Signal transduction & molecular Hematology laboratory, Ludwig Institute for Cancer Research, de Duve Institute, Université catholique de Louvain, Bruxelles, Belgique ; 5 Laboratoire d’hématologie, Inserm, umr866, CHU Dijon, Dijon ; 6 Hématologie, Hôpital Saint-Antoine, Paris ; 7 Hôpital saint-louis, service de biologie cellulaire, Assistance Publique Hôpitaux de Paris, Paris ; 8 Centre d’Investigations Cliniques, Hôpital Saint-Louis, Paris
Introduction. Les néoplasmes myéloprolifératifs (NMP) qui incluent la polyglobulie de Vaquez (PV), la thrombocytémie essentielle (TE) et la myélofibrose primaire (MFP) sont dus à des mutations de JAK2V617F, de la calréticuline (CALRm) et du récepteur à la thrombopoïétine (MPLm). L’interféron α (IFNα) est un traitement qui induit une réponse hématologique chez la majorité des patients ainsi qu’une réponse moléculaire mais très variable selon les patients. Notre objectif est de prédire l’efficacité à long terme de l’IFNα en suivant le comportement des cellules souches hématopoïétiques (CSH) mutées initiatrices de la maladie.
Patients et méthodes. Nous avons réalisé une étude prospective d’une cohorte de 50 patients traités à l’IFNα (40 % TE, 50 % PV et 10 % MFP), dont 34 JAK2V617F, 14 CALRm, deux MPLm. Tous les quatre mois, pour chaque patient, nous avons mesuré les charges alléliques des mutations dans les cellules matures et déterminé l’architecture clonale des mutations au niveau des progéniteurs enrichis en CSH, immatures et engagés.
Nous observons une réponse hématologique d’environ 70 % pour tous les patients. Au cours du suivi, l’IFNα cible les cellules JAK2V617F plus rapidement dans les progéniteurs que dans les cellules matures. Ce ciblage est plus efficace sur les cellules JAK2V617F que les cellules CALRm.
Résultats. Après un suivi médian de 47 mois, 57 % des patients JAK2V617F présentent une réponse moléculaire dans les cellules matures et 82 % dans les progéniteurs. En revanche, après un suivi médian de 35 mois, seulement 20 % des patients CALRm atteignent une réponse moléculaire et identique dans tous les types cellulaires.
Nous avons ensuite développé un modèle mathématique combiné à une analyse bayésienne corrélant bien aux données expérimentales de suivi. Ainsi, nous avons déterminé le comportement et la cinétique de ciblage des CSH mutées par l’IFNα, impossibles à purifier chez l’homme. Dans ce modèle, les CSH mutées s’épuisent lentement par différenciation (après plus d’un an de traitement), ce qui se traduit en conséquence au début du traitement par une augmentation de la fréquence des progéniteurs puis des granulocytes mutés puis par la décroissance voir la disparition des cellules JAK2V617F. Le taux de diminution des CSH JAK2V617F homozygotes est plus important que celui des CSH JAK2V617F hétérozygotes et CALRm et augmente avec la dose d’IFNα (> 100 μg/semaine).
Enfin, le séquençage par NGS des mutations associées ne montre pas d’impact majeur de l’IFNα, hormis chez certains patients l’apparition de mutations DNMT3A ou des variations dans les mutations TET2 et P53.
Conclusion. Au total, nos résultats suggèrent que l’IFNα épuise le compartiment de CSH mutées par mise en différenciation en progéniteurs. Notre étude prédit que l’IFNα peut les éradiquer lentement, mais de façon plus efficace : i) chez les patients homozygotes JAK2V617F par rapport aux hétérozygotes ou bien CALRm, ii) à haute dose d’IFNα et III) chez les patients avec une faible proportion de CSH JAK2V617F hétérozygotes. Ainsi, cette étude permettra de prédire les meilleurs répondeurs au traitement à l’IFNα.
10-04
Diminution du risque thromboembolique et taux élevé de réponses moléculaires complètes dans la polyglobulie de Vaquez sous ropeginterféron α 2b (ropeg) : résultats au long terme d’une étude randomisée
J. Soret-Dulphy1, L. Roy2, J. Rey3, C. Klade4, R. Kralovics5, H. Gisslinger6, B. Cassinat7, JJ. Kiladjian*8
1 Centre d’investigations cliniques, Hôpital Saint-Louis (AP-HP), Paris ; 2 Hématologie, Hôpital Henri Mondor, Créteil ; 3 Hématologie, Institut Paoli-Calmettes, Marseille ; 4 Pharmacologie, AOP Orphan Pharmaceuticals Aktiengesellschaft, Wien, Autriche ; 5 Genetics of hematological disorders, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienne, Autriche ; 6 Department of Hematology and blood coagulation, Medical University of Vienna, Wien, Autriche ; 7 Hôpital saint-louis, service de biologie cellulaire, Assistance Publique Hôpitaux de Paris, Paris ; 8 Centre d’Investigations Cliniques, Hôpital Saint-Louis AP-HP, Université de Paris, Inserm CIC-1427, Paris
Introduction. Les principaux objectifs du traitement de la polyglobulie de Vaquez (PV) sont : prévenir les événements thromboemboliques ; minimiser le risque de progression ; à terme modifier l’histoire naturelle en ciblant sélectivement le clone malin. L’étude PROUD/CONTINUATION-PV a comparé le ropeg au traitement cytoréducteur standard pour les événements thromboemboliques et autres événements indésirables, les réponses hématologiques et moléculaires sur quatre ans.
Patients et méthodes. Les patients (pts) PV (OMS 2008) de ≥ 18 ans n’ayant jamais reçu de cytoréduction ou un traitement court par hydroxyurée (HU) étaient éligibles. 257 pts ont été randomisés pour recevoir du ropeg ou de l’HU pendant 12 mois au cours de l’étude initiale (PROUD-PV). Dans la phase d’extension (CONTINUATION-PV), les pts du groupe HU ont pu changer pour le meilleur traitement disponible. Les évaluations de l’efficacité comprenaient la réponse complète hématologique (RCH) et la réponse complète moléculaire (RCM), selon les critères de l’ELN modifiés. Les pts ayant interrompu le traitement étaient considérés comme non-répondeurs. L’analyse a été effectuée une fois que tous les pts ont atteint 48 mois de traitement ; toutes les données disponibles sur la sécurité ont été incluses.
Résultats. 95 pts dans le groupe ropeg et 76 dans le groupe témoin sont entrés dans la phase d’extension. Au moment de l’analyse, 139 pts étaient toujours à l’étude (74/95 bras ropeg, 65/76 bras contrôle). 97 % des pts du groupe témoin ont continué l’HU. Le taux de RCH était significativement plus élevé dans le groupe ropeg que dans le groupe témoin pendant la quatrième année (60,6 % contre 43,4 % ; p = 0,02). Conformément à ce contrôle efficace des paramètres hématologiques, un taux très faible d’événements indésirables thromboemboliques majeurs a été observé dans le groupe ropeg : 0,0 %, 0,0 % et 1,1 % des patients dans les deuxième, troisième et quatrième années, respectivement. Dans le groupe témoin, ces taux étaient de 0,9 %, 1,4 % et 0,0 %, respectivement. La charge allélique moyenne de JAK2V617F (%V617F) a diminué de 45 % par rapport au taux de départ sur quatre ans dans le groupe ropeg, alors qu’elle a augmenté de 24 % dans le groupe témoin (p < 0,0001). Le taux de réponse moléculaire (partielle ou complète) à 48 mois était significativement plus élevé chez les pts traités par ropeg que dans le groupe témoin (67,0 % contre 25,7 % ; p < 0,0001]). Aucun pt n’a atteint la RCM dans le groupe témoin. Dans le groupe ropeg, 13 pts présentaient 1 % V617F sous le seuil de 1 % au mois 48. En outre, 34 pts du groupe ropeg ont atteint 1 % V617F < 10 % à 48 mois, ce qui laisse espérer que d’autres patients pourraient encore atteindre le seuil < 1 % à l’avenir. Aucun nouveau signal de sécurité n’a été détecté au cours de la quatrième année. Les taux d’effets indésirables liés au traitement étaient semblables dans les deux groupes (ropeg : 28,7 % ; témoin : 22,9 %). Les tumeurs liées à la maladie ou au traitement signalées pendant toute la période de l’étude comprenaient deux cas de leucémie aiguë, deux cas de carcinome basocellulaire et un cas de mélanome malin, tous dans le groupe témoin ; un cas de transformation en myélofibrose a été observé dans chaque groupe.
Conclusion. Le Ropeg au long cours diminue la survenue d’événements thromboemboliques chez les pts porteurs de PV, sans risque leucémogène. Contrairement à l’HU, le ropeg peut induire des réponses moléculaires profondes dont des RCM, soulignant sa capacité à modifier l’histoire naturelle de la maladie. De plus, les pts pourraient obtenant une guérison opérationnelle par ropeg (avec à la fois RCH et RCM), pourraient interrompre le traitement.
10-05
Fédratinib pour le traitement des patients atteints de myélofibrose précédemment traités par ruxolitinib : une réanalyse de l’étude de phase 2 JAKARTA-2
JJ. Kiladjian*1, E. Jourdan2, C. Harrison3, N. Schapp4, AM. Vannucchi5, RT. Silver6, HC. Schouten7, F. Passamonti8, S. Zweegman9
1 Centre d’Investigations Cliniques, Hôpital Saint-Louis, Paris ; 2 Hématologie, CHU, Nîmes ; 3 Hématologie clinique, Guy's and St Thomas’, Londres, Royaume Uni ; 4 Medical centre, Radboud University Nijmegen, Nimègue, Pays-Bas ; 5 University of florence, AOU Careggi, Florence, Italie ; 6 Hematology, Weill Cornell Medical College, New York, États-Unis ; 7 Oncology, University Hospital Maastricht, Maastricht, Pays-Bas ; 8 Hematology, University of Insubria, Varèse, Italie ; 9 Department of Hematology, Amsterdam University Medical Center, VU University Amsterdam, Cancer Center Amsterdam, Amsterdam, Pays-Bas
Introduction. La myélofibrose (MF) est un syndrome myéloprolifératif grave. Les options de traitement (Tx) sont limitées après échec au ruxolitinib (RUX). Le fédratinib (FEDR) est un inhibiteur oral sélectif de JAK2 pour le Tx de la MF intermédiaire-2 ou de haut risque. L’étude JAKARTA-2 a montré une réduction du volume splénique (RVS) ≥ 35 % chez 55 % des patients (pts) évaluables traités par FEDR qui étaient « résistants ou intolérants au RUX » selon l’investigateur et analysée selon la dernière observation reportée (Harrison, Lancet Haematol, 2017).
Patients et méthodes. Nous avons voulu confirmer l’efficacité de FEDR en utilisant des analyses en intention de traiter (ITT) pour tous les pts de JAKARTA-2 et pour un groupe de pts répondant à des critères plus stricts de rechute ou de résistance (R/R), et d’intolérance au RUX (cohorte avec critères stricts [CCS]). Une cohorte d’analyse de sensibilité (CAS) a été réalisée sur un sous-groupe de pts de la CCS qui ont reçu six cycles (C) de FEDR ou arrêté avant C6 pour des raisons autres que l’arrêt de l’étude. Des pts préalablement traités par RUX atteints de MF primitive ou secondaire à une PV ou une TE présentant une rate ≥ 5 cm sous le rebord costal, un ECOG ≤ 2 et des plaquettes ≥ 50 × 109/L ont été inclus. La dose initiale de FEDR était de 400 mg QD en C de 28 jours. Le critère principal était le taux de RVS (TRVS : % de pts avec une RVS ≥ 35 % entre la baseline (BL) et la fin du C6 [EOC6]) évalué par revue centralisée en aveugle. Un critère secondaire était le taux d’amélioration des symptômes (une réduction ≥ 50 % du score total des symptômes [TSS] selon le MFSAF) de BL à EOC6.
Résultats. 97 pts ont été inclus et ont reçu le FEDR (population en ITT). La CCS comprenait 79 pts (81 %) : 65 étaient en R/R au RUX et 14 pts étaient intolérants. Les caractéristiques à l’inclusion et le Tx antérieur par RUX étaient similaires dans la population en ITT, dans la CSS et la CAS. Dans la CCS, l’âge médian était de 66 ans (38-83), 60 % des pts avaient une MF primitive, 23 % une MF post-PV et 18 % une MF post-TE. Le volume médian de la rate à BL était de 2 946 mL (737-7 815) et sa taille médiane de 18 cm (5-36). La durée médiane de Tx antérieur par RUX était de 11,5 mois (1,0-62,4). Les pts ont reçu en médiane 7 C (1-20) de FEDR. Le TRVS dans la CCS était de 30 % (IC95% : 21-42), cohérent avec les résultats de la population en ITT [31 % (IC95% : 21-41)]. Tous les pts avec des données disponibles (n = 41) ont montré un certain degré de RVS à EOC6. À EOC6, le taux d’amélioration des symptômes était de 27 % (IC95% : 17-39) ; 36/44 (82 %) des pts ont présenté une amélioration du TSS. 66 pts répondaient aux critères de la CAS ; Le TRVS était de 36 % (IC95% : 25-49) et le taux d’amélioration des symptômes était de 32 % (IC95% : 21-45). Les événements indésirables (EI) hématologiques de grade ≥ 3 étaient l’anémie (44 %) et la thrombopénie (20 %). Les EI non hématologiques de grade ≥ 3 étaient l’élévation de la lipase (6 %), des ALAT (4 %) et des diarrhées (4 %). Les EI ont conduit à un arrêt de Tx chez 20 % et à des modifications de dose chez 51 % des pts. Six pts sont décédés pendant l’étude ; aucun décès n’a été considéré comme lié au Tx.
Conclusion. Le FEDR a produit des réductions cliniquement significatives de la splénomégalie et des symptômes chez des pts atteints de MF préalablement traités par RUX. Les résultats étaient similaires dans la population en ITT et chez les pts répondant aux critères stricts d’échec au RUX, confirmée en analyse de sensibilité. La tolérance chez ces pts prétraités présentant une maladie avancée était comparable à celle précédemment rapportée.
10-06
Impact pronostique du profil mutationnel dans les syndromes myéloprolifératifs avec thromboses veineuses splanchniques
PE. Debureaux*1, B. Cassinat2, J. Soret-Dulphy1, E. Verger2, N. Maslah2, PE. Rautou3, A. Plessier3, D. Valla3, S. Giraudier2, JJ. Kiladjian1
1 Centre d’investigations cliniques, Hôpital Saint-Louis (AP-HP), Paris ; 2 Service de biologie cellulaire, Hôpital Saint-Louis AP-HP, Paris ; 3 Service d’hépatologie, Hôpital Beaujon AP-HP, Clichy
Introduction. Les thromboses veineuses splanchniques (TVS) sont liées dans 30 à 40 % des cas à la présence d’un syndrome myéloprolifératif (SMP) sous-jacent. Le diagnostic de ces SMP est parfois difficile avec une NFS normale liée à l’hypersplénisme et l’hémodilution des TVS. La découverte des mutations conductrices (JAK2V617F, CALR, MPL) a permis d’en faciliter le diagnostic : JAK2V617F est trouvée dans 80 % de ces SMP, et une mutation de CALR dans 4 % des cas. Les SMP avec TVS (SMP/TVS) sont considérés comme des formes débutantes de SMP au vu de leurs caractéristiques (femme de 40 ans avec une polyglobulie de Vaquez [PV]). Toutefois, la même fréquence d’évolution hématologique chez les PV jeunes au diagnostic (< 45 ans) que ceux plus âgés (> 60 ans) a été observée. L’objectif principal est de chercher des facteurs prédictifs d’évolution hématologique.
Patients et méthodes. Nous avons réalisé une étude rétrospective monocentrique en incluant les patients avec : (1) diagnostic confirmé de TVS, (2) diagnostic de SMP selon la classification OMS 2008, (3) présence d’une mutation conductrice, (4) recherche de mutation additionnelle par NGS. Le panel NGS comprend 36 gènes myéloïdes impliqués dans les SMP. Le critère de jugement principal est l’incidence de transformation en myélofibrose secondaire, leucémie aiguë ou le décès.
Résultats. Sur 200 SMP/SVT référés à notre hôpital, 80 remplissaient les critères d’inclusion. 37 patients (46 %) avaient au moins une mutation additionnelle à la mutation conductrice (mut+). La médiane de suivi est de 11 ans. La majorité des patients sont des femmes (n = 50, 63%) avec une médiane d’âge à 43 ans. Le SMP est une PV chez 52 patients (65 %) et presque tous sont traités par cytoréducteur (94 %) et anticoagulant (99 %). Il n’a pas été observé de différence entre les patients mut+ ou sans mutation additionnelle (mut-) sur les caractéristiques ci-dessus. Les patients mut+ ont un ratio allélique de JAK2V617F supérieur à celui des patients mut- (31 % vs 11 %, p = 0,0001). Les deux classes de mutations additionnelles les plus représentées concernent des gènes impliqués dans la méthylation (31 %) et dans la régulation de la chromatine ou l’épissage (15 %). Il a été observé 10 événements du critère de jugement principal durant le suivi. Les patients mut+ ont eu plus d’événements que les patients mut- (22 % vs 5 %, p = 0,04). L’analyse univariée sur le critère de jugement principal ne retrouve pas d’impact de la présentation clinique ou du type de mutation conductrice. Deux critères moléculaires sont associés à un pronostic défavorable : ratio allélique de JAK2V617F ≥ 50 % (OR 14,7, 95 % IC 3,2-67,9, p = 0,0005) et présence de mutations additionnelles (OR 5,7, 95 % IC 1,11-28,6, p = 0,03), en particulier le sous-groupe d’anomalies touchant la chromatine/épissage (OR 9, 95 % IC 2,1-39, p = 0,003). L’ensemble de ces analyses nous a permis d’identifier un sous-groupe à haut risque d’évolution hématologique défavorable : patients avec ratio allélique de JAK2 ≥ 50 % et/ou anomalie de chromatine/épissage ou Tp53. Les patients à haut risque versus ceux à bas risque ont une diminution de la survie sans événement à 10 ans (81 % vs 100 %, p = 0,001) et de la survie globale à 10 ans (89 % vs 100 %, p = 0,01).
Conclusion. Les SMP/SVT ont un pronostic hétérogène et leur profilage moléculaire permet d’identifier un groupe de haut risque d’événement hématologique dont la prise en charge doit être renforcée. Un traitement de type interféron ou thérapie ciblée, au vu de leur capacité à diminuer la charge allélique des mutations conductrices, pourrait être proposé à ces patients.
10-07
De nouveaux critères clinicobiologiques simples pour identifier les patients à adresser en consultation d’hématologie devant une polyglobulie ?
K. Mahe*1, H. Kerspern2, E. Lippert3, C. Berthou4, F. Couturaud5, JC. Ianotto4
1 Hématologie, CH de Cornouaille Quimper-Concarneau, Quimper ; 2 Laboratoire de biochimie, CHRU de Brest Hôpital Cavale Blanche, Brest ; 3 Laboratoire d’hématologie, CHRU de Brest Hôpital Cavale Blanche, Brest ; 4 Hématologie clinique, CHRU de Brest, hôpital Morvan, Brest ; 5 Médecine interne et pneumologie - ea3878, université de Bretagne occidentale, CHU Brest, Brest
Introduction. Les critères diagnostiques de la polyglobulie (PG) de Vaquez (PV) modifiés en 2016 (classification OMS) avec un abaissement significatif des seuils d’hémoglobine/hématocrite conduisent à une augmentation de suspicion de PV et à la multiplication d’explorations inutiles et consommatrices de temps. L’utilisation de tests simples orientant vers une origine myéloproliférative d’une PG serait intéressante afin de présélectionner les patients à adresser en consultation spécialisée.
La β2-microglobuline (B2m) qui est fréquemment augmentée dans les néoplasies myélo-prolifératives, le taux d’érythropoïétine (EPO) qui est fréquemment abaissé dans les PV et la présence d’un tabagisme sous-jacent (1re cause des PG secondaires) semblent être de bons candidats.
Patients et méthodes. En utilisant une cohorte de patients adressés en hématologie au CHRU pour une PG (observatoire OBENE), nous avons recherché l’intérêt du dosage de la B2m associé ou non à celui de l’EPO et au caractère fumeur ou non du patient pour prédire le diagnostic de PV versus PG secondaire.
Résultats. Une cohorte de 257 patients soit 123 PV et 134 PG secondaires a pu être analysée.
La B2m médiane était plus élevée chez les patients atteints de PV (3,16 vs 1,98 mg/L, p < 0,0001). Ainsi, 83,7 % des PV avaient une B2m augmentée contre 11,9 % chez les PG secondaires.
L’EPO médian était plus bas chez les patients atteints de PV (4,4 vs 12,3 ui/L, p < 0,0001).
Seulement 8 % des patients atteints de PV étaient fumeurs contre 42,6 % chez les PG secondaires (p < 0,0001).
Le fait d’avoir à la fois une B2m non augmentée associé à une EPO non abaissée et à un tabagisme était prédictif de PG secondaire avec une spécificité et une VPP de 100 %, permettant d’exclure une PV. Ceci concerne 16 % des patients de notre cohorte de polyglobulie adressés en consultation spécialisée d’hématologie.
Le fait d’avoir à l’inverse à la fois une B2m augmentée associée à une EPO abaissée et à l’absence de tabagisme était prédictif de PV avec une spécificité et une VPP de 100 %, permettant d’exclure le diagnostic de PG secondaire. Cela concerne 21,4 % des patients de notre cohorte.
Conclusion. Avec ces tests simples, peu coûteux, réalisables en ville, il semble possible de déterminer en amont de la consultation spécialisée les patients à ne pas adresser de manière systématique en consultation d’hématologie sous réserve que ces trois critères : « B2m normale, EPO non abaissée, tabagisme » soient présents. Ce score simple pourrait être réalisé en médecine générale. Ces résultats seraient à reconfirmer sur une plus large cohorte.
10-08
Étude moléculaire de l’évolution clonale d’une mastocytose systémique associée à un syndrome myélodysplasique au cours du traitement : réponse dissociée à la midostaurine et à l’allogreffe
M. Haas*1, F. Llamas-Gutierrez2, ML. Boulland1, M. Roussel1, T. Lamy De La Chapelle3, T. Fest1, R. Houot3, C. Pastoret1
1 Laboratoire d’hématologie, CHU Rennes - Hôpital Pontchaillou, Rennes ; 2 Laboratoire d’anatomopathologie, CHU Rennes - Hôpital Pontchaillou, Rennes ; 3 Service d’hématologie clinique, CHU Rennes - Hôpital Pontchaillou, Rennes
Introduction. La mastocytose systémique (MS) est une maladie hétérogène rare caractérisée par l’accumulation de mastocytes néoplasiques dans divers tissus, principalement la peau et la moelle osseuse. La mutation somatique KIT est une caractéristique diagnostique de la MS et constitue la principale cible thérapeutique dans la mastocytose avancée. Environ 30 % des patients atteints de MS développent simultanément une autre néoplasie hématologique clonale (MS-ANH), associée alors à un mauvais pronostic et à la présence de mutations additionnelles. Nous rapportons un cas de MS associée à un syndrome myélodysplasique (SMD) où la réalisation d’un NGS séquentiel a permis d’évaluer la réponse aux traitements successifs par midostaurine et allogreffe et d’orienter la décision thérapeutique.
Patients et méthodes. Une patiente de 59 ans se présentait en consultation avec une altération de l’état général, une splénomégalie, des sueurs nocturnes et une pancytopénie. Le bilan médullaire identifiait des signes de dysplasie des lignées érythroblastique et granuleuse, associés à la présence de mastocytes dystrophiques en amas exprimant le CD25 et le CD2. La tryptase était élevée à 169 μg/L. Le diagnostic de MS agressive associée à un SMD a été retenu.
Résultats. Au diagnostic, l’analyse moléculaire par NGS retrouvait la mutation KIT p.D816V, et des mutations des gènes TET2, CBL et JAK2. Une première ligne par midostaurine a permis d’obtenir une réponse partielle. La persistance des cytopénies et des signes de dysplasie au myélogramme ont conduit à l’indication d’une allogreffe de cellules souche hématopoïétique (HSCT) permettant d’obtenir une réponse cytologique et moléculaire complète pour le SMD (normalisation de l’hémogramme et disparition de toutes les mutations hormis KIT). Devant la persistance de la tryptase élevée et des mastocytes médullaires à la BOM, un deuxième traitement par midostaurine a été initié permettant d’obtenir une amélioration clinique et la décroissance de la tryptase à 30 μg/L. À 30 mois de l’allogreffe, les cytopénies réapparaissaient et le myélogramme objectivait la réapparition des signes de dysplasie. Le NGS identifiait une mutation TET2 identique à celle du diagnostic associée à une nouvelle mutation du gène SH2B3 confirmant la rechute du SMD.
Discussion. Dans ce cas de MS-ANH, nous avons étudié l’évolution clonale de la maladie par séquençage à haut débit (NGS) au moment du diagnostic et après chaque ligne de traitement par midostaurine et allogreffe. Le profil moléculaire au diagnostic avec cinq mutations dans les gènes KIT, TET2, CBL, et JAK2 est conforme aux données de la littérature. La présence de la mutation KIT p.D816V dans plus de 80 % des cas apporte le rationnel de l’utilisation de la midostaurine en première ligne des MS avancées. Cependant, dans un sous-groupe de patients, l’allogreffe est nécessaire. Des études récentes ont démontré l’efficacité de l’allogreffe dans les MS-ANH, en particulier lorsque la mastocytose est associée à un SMD. Dans notre cas clinique, la HSCT a permis l’obtention d’une rémission moléculaire sur les cibles du SMD (TET2, CBL et JAK2). Cependant, la persistance des critères de mastocytose a conduit à l’utilisation d’une nouvelle ligne de midostaurine. Ce traitement s’est révélé efficace sur la MS mais nous avons observé une rechute clinique et moléculaire du SMD avec la réapparition de la mutation TET2 connue associée à un nouveau variant de SH2B3.
Conclusion. Ce cas clinique illustre la complexité clonale des MS-ANH et l’intérêt de l’analyse moléculaire dans l’évaluation de la réponse au traitement des deux entités.
10-09
Présentation d’une cohorte de patients suivis pour des néoplasies myéloprolifératives non Philadelphie : l’Observatoire du Finistère sud des néoplasies myéloprolifératives (Ofusi)
L. Le Clech*1, M. André2, K. Mahé1, MJ. Rizcallah1, B. Arnaud3, JC. Ianotto4, P. Hutin1, R. Le Calloch1
1 Médecine interne, maladies infectieuses et du sang, Hôpital Laënnec, CHIC Quimper, Quimper ; 2 Centre d’investigation clinique, Hôpital Laënnec, CHIC Quimper, Quimper ; 3 Laboratoire d’hématologie, Hôpital Laënnec, CHIC Quimper, Quimper ; 4 Hématologie clinique, CHRU de Brest, hôpital Morvan, Brest
Introduction. Les néoplasies myéloprolifératives non Philadelphie (NMP Phi-) sont des maladies rares mais d’évolution chronique. Les risques engendrés par ces pathologies sont principalement les événements thrombotiques ou hémorragiques ainsi que la transformation vers une hémopathie plus grave. Nous souhaitons vous présenter ici les premières descriptions d’une cohorte de NMP Phi- nommée Ofusi.
Patients et méthodes. Cet observatoire ouvert depuis novembre 2018 recense tous les patients suivis pour un NMP Phi- dans notre centre hospitalier ainsi que sur le groupement territorial du Finistère sud (GHT n̊2). Les patients sont inclus après recueil de leur non-opposition et remise d’une lettre d’information.
Les données recueillies concernent le patient (âge, sexe, antécédents, facteurs de risque cardio-vasculaires) et la pathologie (caractéristiques biologiques du SMP : sang, moelle, biologie moléculaire). Les événements principaux du suivi sont également relevés : traitement cytoréducteur, traitement antiagrégant ou anticoagulant, événements thrombotiques, néoplasies, taux de transformation et décès.
Résultats. Après un an de recueil, les données de 168 patients ont pu être collectées : 94 (56 %) femmes et 74 (44 %) hommes d’âge moyen de 65 ans (17 ; 93 ans). Le suivi médian est de cinq ans (3-9 ans). Le diagnostic réalisé était 94 (56 %) thrombocytémies essentielles, 52 (31 %) polyglobulies de Vaquez, 15 (9 %) myélofibroses primitives, et sept (0,4 %) SMP inclassables. La mutation du gène Jak2 était la plus fréquente (n = 116, 69% V617F et exon 12), suivie de calréticuline (n = 18), mutation inconnue ou en cours (n = 15), triple négative (n = 14) et MPL (n = 3).
Cent trois (61 %) patients présentaient un haut risque vasculaire au diagnostic dont 36 patients avaient eu un antécédent thrombotique antérieur. 15 (9 %) ont présenté un nouvel événement au diagnostic (thrombose veineuse n = 4/thrombose artérielle n = 11).
Les patients ont reçu en moyenne 1,4 ligne de traitement cytoréducteur, majoritairement de l’Hydréa (n = 92). Les indications de changement de traitement (n = 40) étaient une intolérance (n = 18) puis une inefficacité du traitement (n = 10).
Dix-huit (10,7 %) patients ont présenté une transformation de leur maladie en myélofibrose, myélodysplasie ou leucémie aiguë et 27 (16 %) ont eu un diagnostic d’une autre néoplasie dans leur suivi (24 tumeurs solides, trois hémopathies lymphoïdes).
Conclusion. Cette base de données est évolutive de manière prospective, par l’inclusion de nouveaux patients et leur suivi longitudinal. Elle fait miroir à la cohorte du Finistère Nord, OBENE, permettant un travail collaboratif au sein de la Fédération interhospitalière d’hématologie de Bretagne Occidentale (FIHBO). Cette association doit permettre la réalisation de nombreuses études ayant pour objectifs d’améliorer la prise en charge des patients et de mettre éventuellement en avant des risques évolutifs méconnus. Une collection biologique de recherche est également en cours afin de répondre à des projets nationaux du FIM (France-Intergroupe des syndromes Myéloprolifératifs).
10-10
Outils utilisés en vie réelle pour le diagnostic de 342 polyglobulies de Vaquez
JM. Bons1, O. Fitoussi*2, C. Lenoir2, M. Boisseau3, S. Bologna4, AV. Doncker5, B. Bareau5, E. Legouffe6, M. Cliquennois7, M. Balhadère2, C. Dauriac8
1 Hématologie, Centre de Radiothérapie Joseph Bellot, Montluçon ; 2 Hématologie, Polyclinique Bordeaux Nord Aquitaine, Bordeaux ; 3 Hématologie, Clinique Pasteur, Toulouse ; 4 Oncohématologie, Sel Du Centre D’oncologie de Gentilly, Nancy ; 5 Service d’hématologie et médecine interne, Hôpital privé Sévigné, Vivalto Santé, Cesson-Sévigné ; 6 Hématologie, Oncogard, centre hospitalier du Gard, Alès ; 7 Hématologie, Hôpital privé de Villeneuve d’Ascq - Ramsay Santé, Villeneuve-d’Ascq ; 8 Oncohématologie, clinique Victor Hugo, Institut interrégional de cancérologie, Le Mans
Introduction. Les syndromes myéloprolifératifs (SMP) occupent une part importante de l’activité des différents centres du secteur libéral hématologique français, réunis au sein du réseau HLA (hématologues libéraux associés). Il nous est apparu intéressant de colliger tous les éléments utilisés par les centres HLA pour le diagnostic de polyglobulie de Vaquez (PV), dans des conditions de vie réelle.
Patients et méthodes. Quinze centres HLA ont enregistré, grâce à un eCRF spécifique, sur une période de six mois (du 5 novembre 2018 au 5 mai 2019), les éléments diagnostiques de 342 patients atteints de PV. Le temps médian entre le diagnostic et la saisie des données dans l’eCRF était de cinq ans, ce qui signifie que le diagnostic de PV pouvait être récent comme ancien (jusqu’à 31 ans). Les critères de diagnostic retenus pour les PV entre 2008 et 2016 étaient ceux de la WHO 2008 et ceux de la classification OMS 2016 pour les PV depuis 2016.
Résultats. L’âge moyen de la population est de 71 ans (34 à 99 ans), avec 64 % d’hommes et 36 % de femmes. Au diagnostic, 100 % des patients ont eu un hémogramme ; 90 % soit 307 patients une recherche de la mutation du JAK2V617F (86 % mutée), 16 patients une recherche de la mutation de l’exon 12 (25 % positive) ; 40 % une masse sanguine ; 74 % un dosage d’EPO endogène ; 44 % une biopsie ostéomédullaire (BOM) ; 7 % une étude de pousse spontanée des progéniteurs érythrocytaires in vitro.
Discussion. Depuis la classification OMS 2001, les critères diagnostiques de PV sont en constante évolution ; ainsi, suite à la découverte en 2005 de la mutation du JAK2V617F et en 2007 de la mutation de l’exon 12 et de leurs implications diagnostiques dans les SMP, les critères WHO 2008 ont intégré en critère majeur ce statut mutationnel. Si l’on applique les critères diagnostiques WHO 2008, sur les 118 patients de cette étude HLA, diagnostiqués entre 2008 et 2016, seuls 55 % répondent à ces critères. Maintenant, si l’on considère les critères actuels OMS 2016, sur les 166 patients diagnostiqués depuis 2016, seuls 34 % ont validé ces critères diagnostiques, essentiellement par défaut de BOM. Avant 2016, la possibilité d’avoir recours à un critère mineur tel que la pousse des progéniteurs érythrocytaires in vitro, par ailleurs de réalisation et de standardisation difficiles, permettait, en vie réelle, d’éviter, aux patients âgés ou fragiles, la pratique d’une BOM de qualité parfois médiocre.
Conclusion. Les critères diagnostiques exigés OMS 2016 de PV, en réduisant le nombre de critères exigés, ont considérablement simplifié, en vie réelle, le parcours diagnostic des patients atteints de PV ; et ce, d’autant plus que la BOM n’est pas obligatoire si l’hémoglobine et/ou l’hématocrite initiales atteignent des valeurs élevées qui étaient déjà utilisées comme critère majeur dans la classification WHO 2008.
10-11
SYTHRON (SYndrome myéloprolifératif, THROmbose et hémogramme Normal) : une étude de l’Intergroupe des syndromes myéloprolifératifs (FIM)
Y. Le Bris*1, JC. Chomel2, K. Laribi3, C. Guénot4, P. Sujobert5, G. Denis6, L. Legros7, C. James8, O. Mansier8, M. Loirat9, S. Guillou10, P. Etancelin11, S. Tavitian12, E. Delabesse13, S. Vigouroux14, B. Dupriez15, S. Natarajan-Amé16, L. Miguet17, V. Dubruille18, M. Artifoni19, P. Peterlin18, J. Graveleau20, C. Ternisien1, E. Lippert21, T. Eugene22, J. Rey23, V. Ugo24, JJ. Kiladjian25, P. Cony-Makhoul26, C. Pastoret27, D. Naguib28, B. Burroni29, MC. Béné1, M. Fouassier1, JC. Ianotto30
1 Service d’hématologie biologique, Hôtel-Dieu, Nantes ; 2 Département de biologie moléculaire, Université de Poitiers, Poitiers ; 3 Hématologie clinique, CH Le Mans, Le Mans ; 4 Hématologie biologie, CH Le Mans, Le Mans ; 5 Hématologie, CH Lyon Sud, Pierre-Bénite ; 6 Médecine interne et hématologie, CH de Rochefort, Rochefort ; 7 Hématologie clinique, Groupe hospitalier l’Archet, Nice ; 8 Laboratoire d’hématologie, Hôpital Haut Lévêque, Pessac ; 9 Service d’hématologie, Hôpital Saint Nazaire, Saint-Nazaire ; 10 Laboratoire d’hématologie, CHU Reims, Reims ; 11 Laboratoire de Génétique Oncologique, Centre Henri Becquerel, ROUEN ; 12 Hématologie clinique, IUCT Oncopole, Toulouse ; 13 Hématologie biologie, IUCT Oncopole, Toulouse ; 14 Hématologie, CH Départemental Vendée, La Roche-sur-Yon ; 15 Hématologie clinique, CH de Lens, Lens ; 16 Service d’hématologie clinique, CHU Strasbourg, Strasbourg ; 17 Laboratoire d’hématologie, CHRU Hôpitaux Universitaires Strasbourg, Strasbourg ; 18 Service d’hématologie clinique, Hôtel-Dieu, Nantes ; 19 Médecine vasculaire, CHU Nantes, Nantes ; 20 Hématologie, CH de Saint Nazaire, Saint-Nazaire ; 21 Laboratoire d’hématologie, CHU de Bordeaux, Bordeaux ; 22 Médecine nucléaire, CHU Hôtel-Dieu, Place Alexis-Ricordeau, Nantes, France, Nantes ; 23 Hématologie clinique, Institut Paoli-Calmettes, Marseille ; 24 Laboratoire d’hématologie, CHRU de Brest, Brest ; 25 Centre d’Investigations Cliniques, Hôpital Saint-Louis, Paris ; 26 Centre de recherche clinique, CH Annecy Genevois, Metz-Tessy ; 27 Laboratoire d’Hématologie, CHU de Rennes, Rennes ; 28 Laboratoire d’Hématologie, CHU Côte de Nacre, Caen ; 29 Laboratoire de pathologie, Hôpital Cochin, Paris ; 30 Hématologie clinique, CHRU de Brest, Brest
Introduction. La thrombose est la principale cause de morbidité des néoplasies myéloprolifératives (NMP). Sa prise en charge repose sur la mise en place d’un traitement anti-thrombotique et dans certains cas d’un agent cytoréducteur (ACR). En cas de thrombose atypique, idiopathique ou récidivante chez un sujet jeune, un diagnostic de NMP doit être recherché même en l’absence de signe de prolifération sur l’hémogramme. En l’absence de myéloprolifération, l’utilisation d’un ACR pour réduire le risque de récidive de la thrombose ou d’évolution hématologique est empirique.
Ce travail se propose de recueillir les cas de thrombose artérielle ou veineuse associée à une NMP mais avec un hémogramme normal au diagnostic. L’objectif est de mesurer l’incidence de récidive thrombotique et d’évolution hématologique. L’impact des traitements par ACR, anticoagulant et/ou antiagrégant plaquettaire sera ainsi évalué.
Patients et méthodes. Il s’agit d’une étude de cohorte observationnelle avec 200 patients attendus. L’inclusion est multicentrique rétrospective et prospective selon les critères suivants :
– diagnostic d’un accident thrombotique artériel ou veineux profond,
– diagnostic d’une NMP caractérisée par la détection d’une anomalie moléculaire (mutation JAK2, CALR ou MPL) et/ou d’une anomalie à la biopsie ostéomédullaire en faveur d’une NMP,
– hémogramme non évocateur d’une polyglobulie de Vaquez, d’une thrombocytémie essentielle ou d’une myélofibrose (seuils OMS 2016) au moment de l’évènement thrombotique.
Résultats. L’ouverture des inclusions a eu lieu le 2 juillet 2019. Sur les 32 centres qui ont accepté de participer à l’étude, le bilan à six mois arrêté le 2 janvier 2020 montre que 17 centres ont inclus 79 patients (H/F = 42/37). L’âge médian au moment du diagnostic de NMP est de 56 ans (23-91). La thrombose inaugurale concerne un territoire soit digestif (52 %), soit cérébral (28 %) soit un autre territoire (20 %). Le diagnostic de NMP a pu être objectivé par la mise en évidence d’une mutation JAK2V617F chez tous les patients, associée à, lorsque cela a été exploré, des anomalies sur la BOM (52 %), une pousse spontanée des progéniteurs (57 %) et/ou une masse sanguine élevée (20 %). Un traitement cytoréducteur a été mis en place chez 53 % des patients. Un suivi clinicobiologique des patients inclus a été renseigné pour 57 d’entre eux (70 %).
Conclusion. Ces premiers résultats démontrent la faisabilité de l’étude et, notamment, que l’objectif de 200 patients est atteignable en moins de deux ans. La localisation de la thrombose est variable en cas de NMP avec hémogramme sans signe de myéloprolifération. Tous les patients de cette série présentent une mutation JAK2V617F. Les caractéristiques des patients traités par ACR et leur impact sur la récidive thrombotique et l’évolution hématologique seront évalués. Les centres qui souhaitent participer et inclure sont les bienvenus.
10-12
Une faible charge allélique JAK2V617F pourrait aider à identifier de nouveaux sous-groupes de polyglobulie de Vaquez et de thrombocytémie essentielle
C. Croizier*1, B. Lebecque2, T. Tassin3, W. Bombardier3, AP. Grandjean4, AM. Milesi-Lecat5, B. Aubin6, V. Pante7, S. Trouillier8, JB. Bouillon-Minois9, R. Martel10, N. Boiret-Dupré3, N. Tribalat3, E. Louis3, JO. Bay11, M. Berger12, C. Bourgne13
1 Hématologie clinique et thérapie cellulaire, CHU Estaing, Clermont-Ferrand ; 2 Laboratoire d’hématologie, CHU Estaing, Clermont-Ferrand, EA 7453 CHELTER, Université Clermont Auvergne ; 3 Hématologie biologique, CHU Estaing, Clermont-Ferrand ; 4 Oncologie médicale, CH de Montluçon, Montluçon ; 5 Médecine interne, Jacques Lacarin, Vichy ; 6 Hématologie clinique, CH de Moulins-Yzeure, Moulins ; 7 Oncologie médicale, CH Emile Roux, Le Puy-en-Velay ; 8 Médecine interne, CH Aurillac, Aurillac ; 9 Urgences Samu Smur, CHU Gabriel-Montpied, CNRS UMR 6024 LAPSCO, Clermont-Ferrand ; 10 Urgences Samu Smur, CH Emile Roux, Le Puy-en-Velay ; 11 Hématologie clinique et thérapie cellulaire/EA Creat 7283, CHU Estaing, Clermont-Ferrand, France, Clermont-Ferrand ; 12 Hématologie biologique, CHU Estaing, Clermont-Ferrand ; 13 Hématologie biologique, CHU Clermont-Ferrand, Clermont-Ferrand
Introduction. La mutation JAK2V617F constitue un critère majeur dans le diagnostic des syndromes myéloprolifératifs (SMP). Cependant, l’impact clinicobiologique d’une faible charge allélique JAK2V617F reste discuté. Nous avons évalué de façon rétrospective la valeur diagnostique de ce paramètre pour les SMP de type thrombocytémie essentielle (TE) et Polyglobulie de Vaquez (PV) sur une période de 12 années en Auvergne.
Résultats. Entre le 5 février 2007 et le 21 août 2019, 6 258 patients ont bénéficié d’une recherche de mutation JAK2V617F. Quatre cents résultats (6,39 %) ressortent faiblement positifs (< 4%) ; 21 (5,25%) PV ont été diagnostiqués et 46 (11,5 %) TE. Dans le reste de la cohorte, 16 patients (4 %) ont un diagnostic de myélofibrose primitive, 22 (5,5 %) de fausses polyglobulies, 25 (6,25 %) de polyglobulies secondaires et 90 (22,5 %) pour lequel nous avons un manque d’information.
Dans le groupe PV (âge médian 63 ans (44-85), sex-ratio H/F 3,2), la charge allélique mutée médiane est de 0,89 % (0,04-3,9). Aucun patient n’a de mutation JAK2 exon12. Les taux médians d’hémoglobine (Hb) sont de 17,5 g/dL (14,3-24,2), d’hématocrite (Hte) à 51,1 % (43,8-65,1) et de plaquettes (Plq) à 197 G/L (101-660). Le taux d’érythropoïétine (EPO) plasmatique médian est de 5,3 mUI/mL (2,2-19,9). Le diagnostic a été appuyé par l’analyse de la biopsie ostéomédullaire (BOM) (7/11, soit 63,6 %) et/ou l’augmentation du VGT (4/11, soit 36,4 %).
Les patients PV ayant un taux de mutation JAK2V617F > 4 % (n = 21 ; âge médian 74 ans (41-94) ; H/F 1,3 ; JAK2V617F médian 53,5 % (24,7-86,8)), ont des taux d’Hte (médiane 54 % ; p = 0,01) et de Plq (médiane 588 G/L ; p = 8,7 × 10-7) significativement plus élevés et un taux d’EPO plasmatique plus bas (médiane 1,6 mUI/mL ; p = 2,4 × 10-6).
Dans le groupe TE (âge médian 68,5 ans (32-93), H/F 0,76), la charge allélique mutée médiane est de 0,33 % (0,01-3,4). Le taux de Plq moyen est de 785,4 G/L (400-1 600). La recherche des mutations CALR et MPL (40/46 patients) a identifié une mutation CALR chez 18 patients (45 %) et MPL chez quatre patients (10 %). Pour 13/16 patients l’analyse de la BOM a appuyé le diagnostic de TE.
En comparaison avec le groupe (n = 46) de TE ayant un taux de mutation JAK2V617F > 4% (médiane 22,5 % (11,5-52,1)), aucune différence significative n’a été observé sur les paramètres sanguins, avec notamment un taux de Plq médian de 706 G/L (311-1558) (p = 0,4).
Conclusion. Cette étude rétrospective a montré qu’un taux faible de mutation JAK2V617F peut être retrouvé dans diverses hémopathies (6,39 % de notre cohorte), et permettre le diagnostic d’un SMP (16,8 % des cas) en intégrant les autres critères définis par l’OMS. Aucune mutation JAK2 exon 12 n’a été observée dans les PV JAK2V617F faible, alors qu’une proportion élevée de patients présentent une mutation CALR ou MPL (48 %) dans le groupe TE JAK2V617F faible. À l’heure où les techniques de biologie moléculaire évoluent (PCR digitale), il paraît nécessaire de mieux définir les sous-groupes PV JAK2 faible, TE JAK2 faible avec ou sans mutation CALR ou MPL sur une cohorte plus importante.
10-13
Étude de phase 2 portant sur le luspatercept chez des patients souffrant d’anémie associée à la myélofibrose
JJ. Kiladjian*1, AM. Vannucchi2, F. Passamonti3, M. Kremyanskaya4, J. Gotlib5, JM. Palmer6, K. Mccaul7, V. Ribrag8, A. Mead9, C. Harrison10, R. Mesa11, G. Barosi12, TG. Gerike13, S. Verstovsek14, AT. Gerds15
1 Centre d’Investigations Cliniques, Hôpital Saint-Louis et Université Paris Diderot, Paris ; 2 Centre de recherche et d’innovation sur les néoplasies myéloprolifératives, AOU Careggi, Florence, Italie ; 3 Département de médecine et chirurgie, Université d’Insubria, Varese, Italie ; 4 Division of Hematology and Medical Oncology, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai Hospital, New York, NY, États-Unis ; 5 Hematology division, Stanford Cancer Institute, Stanford, CA, États-Unis ; 6 Division of Hematology/Oncology, Mayo Clinic, Phoenix, AZ, États-Unis ; 7 Department of Hematology and bone marrow transplant, Avera Cancer Institute, Sioux Falls, SD, États-Unis ; 8 Hématologie, Institut Gustave Roussy, Villejuif ; 9 Department of Haematology, Oxford University Hospitals NHS Foundation Trust, Oxford, Royaume Uni ; 10 Hématologie clinique, Guy's and St Thomas’ NHS Foundation Trust, Londres, Royaume Uni ; 11 Mays cancer Center, UT Health San Antonio Cancer Center, San Antonio, TX, États-Unis ; 12 Laboratorio di epidemiologia clinica, Fondazione IRCCS Policlinico San Matteo, Pavia, Italie ; 13 Clinical research & development, Bristol-Myers Squibb, Summit, NJ, États-Unis ; 14 Department of leukemia, The University of Texas MD Anderson Cancer Center, Houston, TX, États-Unis ; 15 Cleveland Clinic, Taussig Cancer Institute, Cleveland, OH, États-Unis
Introduction. Nous rapportons ici les résultats intermédiaires de l’étude ouverte de phase 2 toujours en cours portant sur le luspatercept chez des patients atteints de myélofibrose (MF) et d’anémie.
Patients et méthodes. 76 patients atteints de MF et d’anémie ont été recrutés, dont 43 ne recevant pas de ruxolitinib lors de leur inclusion dans l’étude et ayant reçu soit aucune transfusion de GR (n = 22 ; groupe 1), soit deux à quatre unités de GR (UGR)/28 jours au cours des 12 semaines (sem) précédant le traitement (n = 21 ; groupe 2). 33 patients recrutés recevaient une dose journalière stable de ruxolitinib depuis ≥ 16 sem ; 14 ne recevaient pas de transfusions de GR (groupe 3A) et 19 en recevaient (groupe 3B).
Les patients ont reçu 1,0 à 1,75 mg/kg de luspatercept tous les 21 jours. Le critère d’évaluation principal a été évalué au cours des 24 premières sem de l’étude ; pour les groupes 1 et 3A, il s’agissait d’une augmentation ≥ 1,5 g/L de l’hémoglobine par rapport à sa valeur initiale lors de chaque évaluation pendant ≥ 12 sem consécutives ; pour les groupes 2 et 3B, le critère d’évaluation principal était l’indépendance vis-à-vis des transfusions de GR (ITGR) pendant ≥ 12 sem consécutives. Les critères d’évaluation secondaires étaient la sécurité, une augmentation moyenne de Hb ≥ 1,5 g/dL (groupes 1 et 3A) et une réduction ≥ 50 % du nombre de transfusions de GR avec une réduction d’au moins quatre UGR (groupes 2 et 3B), ces deux réponses durant ≥ 2 sem consécutives, dans les 24 sem suivant l’inclusion dans l’étude. Les données de projet thérapeutique ont été analysées à la date du 5 août 2019.
Résultats. 3/22 (14 %) et 3/14 (21 %) patients des groupes 1 et 3A, respectivement, sont parvenus à une augmentation ≥ 1,5 g/dL de Hb par rapport à sa valeur initiale à chaque mesure sur toute période de 12 sem consécutives. 2/21 (10 %) et 6/19 (32 %) patients des groupes 2 et 3B, respectivement, sont parvenus à l’ITGR sur toute période de 12 sem consécutives. La durée médiane de l’ITGR a été de 32 sem (intervalle 16-49 sem) pour le groupe 2 et de 39 sem (intervalle 12-77 sem) pour le groupe 3B. Quatre (18 %) et neuf (64 %) patients des groupes 1 et 3A sont parvenus à une augmentation moyenne de Hb ≥ 1,5 g/dL. Huit (38 %) et 10 (53 %) patients des groupes 2 et 3B sont parvenus à une réduction ≥ 50 % par rapport à sa valeur initiale du nombre de transfusions de GR.
Les effets indésirables les plus fréquents associés au traitement étaient hypertension (12 % de patients), douleurs osseuses (9 %) et diarrhée (5 %). Trois événements associés au traitement de grade 3 ont été rapportés (hypertension, diarrhée et déshydratation).
Conclusion. Les résultats initiaux de cette étude en cours suggèrent une activité clinique du luspatercept chez les patients souffrant d’anémie associée à la myélofibrose.
10-14
Le polymorphisme de LNK (SH2B3) rs3184504 est associé aux syndromes myéloprolifératifs avec mutation de CALR
D. Luque Paz*1, F. Boyer2, JC. Ianotto3, R. Jouanneau-Courville1, J. Riou4, A. Chauveau5, K. Laribi6, V. Cussac7, A. Blouet8, M. Truchan-Graczyk9, L. Cottin1, N. Jézéquel10, A. Delisle1, C. Orvain11, M. Hunault-Berger12, E. Lippert5, V. Ugo1, L. Velazquez13
1 Laboratoire d’hématologie, CHU - CHU Angers, Angers ; 2 Maladies du sang, CHU - CHU Angers, Angers ; 3 Hématologie clinique, CHRU de Brest, hôpital Morvan, Brest ; 4 Université d’angers, Inserm 1066 MINT, Angers ; 5 Laboratoire d’hématologie, CHRU de Brest, Brest ; 6 Hématologie, centre hospitalier, Le Mans ; 7 Laboratoire d’hématologie, CH - Le Mans, Le Mans ; 8 Oncohématologie, CH de Cholet, Cholet ; 9 Hématologie, CENTRE HOSPITALIER DE SAUMUR, Saumur ; 10 Laboratoire d’hématologie, CHRU Cavale Blanche, Brest ; 11 Hématologie, CHU d’Angers, Angers ; 12 Service des maladies du sang, CHU - CHU Angers, Angers ; 13 Institut André lwoff, Inserm UMRS-MD1197, Bobigny
Introduction. Une mutation promotrice des gènes JAK2, CALR ou MPL, à l’origine d’une dérégulation de la voie de signalisation JAK2-STAT5, est retrouvée dans plus 80 % des cas de syndromes myéloprolifératifs non-LMC (SMP). Les mutations de CALR sont retrouvées dans 15 à 25 % des cas de thrombocytémie essentielle et de myélofibrose primitive JAK2 non mutés. Des facteurs de prédisposition génétique aux syndromes myéloprolifératifs ont été décrits : certains sont rares mais associés à une forte pénétrance et à des formes familiales, comme la duplication ATG2P/GSKIP, d’autres sont fréquents dans la population générale, comme l’haplotype JAK2 46/1. Une association entre le polymorphisme du gène SH2B3 (c.784T>C, allèle T, p.W262R) codant LNK, un régulateur négatif de la voie JAK2-STAT5, et les syndromes myéloprolifératifs avec mutation de JAK2 a été précédemment décrite (Lesteven et al. Blood 2014). L’objectif de ce travail était d’étudier l’association entre ce polymorphisme et les syndromes myéloprolifératifs avec mutation de CALR.
Patients et méthodes. 139 patients atteints d’un syndrome myéloprolifératif (105 TE, 31 MFP et trois inclassables) avec mutation de CALR (61 types 1, 39 types 2 et 39 autres types) ont été inclus. Le génotype de SH2B3/LNK (présence allèles T/C) a été obtenu par séquençage NGS ou par qPCR Taqman spécifique à partir d’ADN dérivé des leucocytes sanguins. Pour comparaison, la fréquence du polymorphisme a été recueillie sur la base de données GNOMAD (population générale européenne NFE, n = 57520) et dans l’article Lesteven et al Blood 2014 ; 123: p794-5 (SMP avec mutation JAK2, n = 184). Les associations ont été mesurées par des odds ratio et les comparaisons des données faites par des tests non paramétriques (tests de Fisher ou de Mann-Whitney, logiciel R http://www.R-project.org, Austria).
Résultats. Dans cette cohorte nous avons observé un génotype TT chez 39 patients, TC chez 76 et CC chez 24, ce qui correspond à une fréquence de l’allèle T de 55,4 %. Cette fréquence est de 48,7 % dans la population générale et de 56,8 % dans les SMP mutés JAK2. Une association significative est retrouvée entre l’allèle T (W262) et les syndromes myéloprolifératifs mutés CALR (OR = 1,3 [1,02-1,7] ; p = 0,03). Nous avons observé une légère surreprésentation du génotype TT dans la myélofibrose primitive par rapport à la thrombocytémie essentielle (42 % vs 25 %, p = 0,07). Aucune différence n’a été retrouvée entre les différents génotypes en termes de répartition des types de mutation de CALR ou de charge allélique. Nous avons enfin comparé les caractéristiques clinicobiologiques des TE homozygotes TT (n = 26) aux TE homozygotes CC (n = 18) et les génotypes TT présentaient un âge plus jeune au diagnostic (56 vs 65 ans, p = 0,013).
Conclusion. Ce travail montre que le polymorphisme de SH2B3/LNK rs3184504 (allèle T), décrit comme associé aux SMP avec mutation de JAK2, est également associé aux SMP avec mutation de CALR.
10-15
MPN personalized risk calcultator et son usage en vie réelle appliqué à une population de sujets jeunes thrombocytémie essentielle
V. Rebiere*1, JJ. Tudesq2, E. Lippert3, M. Darmon4, JC. Ianotto5
1 Hématologie Clinique, CHRU de Brest, Avenue Foch, Brest, France, Brest ; 2 Réanimation médicale, AP-HP Hôpital Saint-Louis, PARIS ; 3 Laboratoire d’hématologie, CHU de Brest, Brest ; 4 Réanimation médicale, Hôpital Saint-Louis, Paris ; 5 Hématologie clinique, CHRU de Brest, hôpital Morvan, Brest
Introduction. Les néoplasies myéloprolifératives (NMP) sont des pathologies clonales chroniques identifiées par une ou des proliférations sanguines secondaires à l’acquisition d’une mutation activatrice. Les principales NPM sont la polyglobulie de Vaquez (PV), la thrombocytémie essentielle (TE) et la myélofibrose primitive (MF). La prise en charge thérapeutique associe souvent un cytoréducteur (dont l’interféron-pégylé ou l’Hydroxyurée ou l’Anagrélide) à de l’aspirine à faible dose. Elle est prescrite afin de réduire le risque de thromboses (artérielles ou veineuses) tout en limitant le risque de transformation vers des phases accélérées (myélofibrose secondaire ou leucémie aiguë myéloïde). Des progrès majeurs ont été réalisés au cours des dix dernières années en matière de génomique et de compréhension de l’évolution clonale dans les NPM. La première utilisation concrète de ces progrès a été de proposer un nouveau score dit MPN Personalized Risk Calculator fin 2018. Pour les patients en phase chronique, ce score intégratif (clinico-biologico-moléculaire) se propose de prédire la survie sans événement (sans transformation) et la survie globale à cinq et 10 ans. L’idée de ce travail est d’évaluer la pertinence de cette approche personnalisée dans une population particulière de patients (patients avec TE de moins de 60 ans) pour qui la prise en charge thérapeutique repose surtout sur la limitation des prescriptions à la seule aspirine du fait du risque évolutif très faible.
Patients et méthodes. Ce score a été appliqué avec l’ensemble des données cliniques, biologiques et moléculaires disponibles (JAK2, CALR, MPL) de notre cohorte de 165 patients TE de moins de 60 ans.
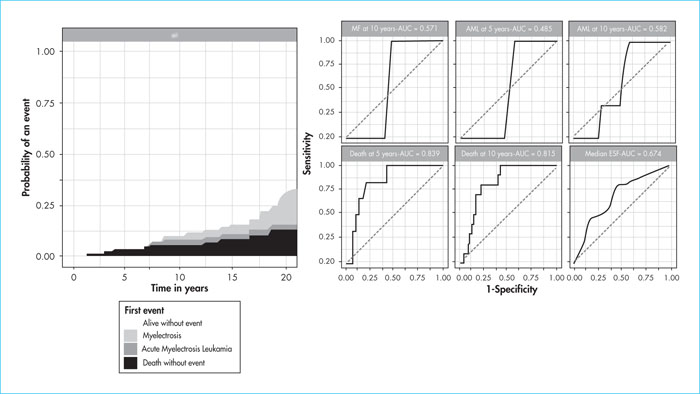
Résultats. Voir figure 1.

Conclusion. Il s’agit à notre connaissance de la première utilisation en vie réelle de ce calculateur avec une performance de concordance globale intéressante sur les paramètres de transformation en MF, LAM et décès, nécessitant d’être vérifiée de manière prospective.
10-16
L’expression des mutations drivers des syndromes myéloprolifératifs favorise la production d’un transcrit proleucémogène de RUNX1
M. Hautin-Ropert*1, T. Bergot1, O. Mansier2, J. Delahousse3, A. Paci3, L. Corcos1, D. Bernard1, E. Lippert4
1 Équipe Ecla, Inserm UMR 1078, Brest ; 2 Laboratoire d’hématologie, CHU de Bordeaux, Bordeaux ; 3 Département de biologie-pathologie, Gustave Roussy, Rue Edouard Vaillant, Villejuif, France, Villejuif ; 4 Service d’hématologie-biologique, CHRU Brest, Brest
Introduction. Les anomalies de l’épissage font partie des mécanismes clés de leucémogenèse et cancérogenèse. Elles sont parfois liées à la présence de mutations dans les gènes du spliceosome ou au niveau de sites d’épissage. Cependant, même en l’absence de mutation affectant la machinerie d’épissage, des analyses transcriptomiques ont montré des différences d’épissage entre les cellules de leucémies aiguës myéloblastiques (LAM) et les cellules normales.
Les anomalies de l’épissage sont peu étudiées dans les syndromes myéloprolifératifs (SMP). Ces hémopathies malignes chroniques sont caractérisées par une prolifération aberrante des progéniteurs myéloïdes, liées à des mutations drivers touchant les gènes JAK2, CALR ou MPL. Ces dernières sont responsables d’une hypersensibilité aux cytokines et d’une activation constitutive des voies de signalisation, notamment la voie JAK2-STAT5. Ces pathologies peuvent évoluer de manière imprévisible en LAM. Notre équipe souhaite caractériser les anomalies d’épissage survenant suite à l’expression de mutations communément décrites dans les SMP, et d’en comprendre les mécanismes d’apparition.
Matériels et méthodes. Un modèle d’expression inductible des mutations drivers de JAK2 (JAK2V617F) ou de CALR (CALRdel52 et CALRins5), a été généré par transfection lentivirale d’une lignée hématopoïétique dépendante des cytokines (UT-7). Pour valider l’impact fonctionnel des mutations, nous avons testé la prolifération cellulaire en milieu dépourvu de cytokines, ainsi que la phosphorylation de STAT5. L’étude de l’épissage a été faite par RT-qPCR spécifique de transcrits pour six gènes importants dans la leucémogenèse. Un modèle double-mutant (DM) combinant la mutation JAK2V617F en association avec la mutation IDH2R140Q a également été généré. Le dosage du 2-hydroxyglutarate (2-HG) a permis de valider l’impact fonctionnel de cette mutation.
Résultats. Les cellules exprimant les mutations drivers acquièrent une indépendance aux cytokines pour leur croissance, avec un temps de doublement équivalent à celui des cellules cultivées en présence de cytokines (entre 48 et 72 h). Elles présentent également une phosphorylation spontanée de STAT5, témoin d’une activation constitutive de la voie JAK2-STAT5. Concernant l’épissage, nous avons pu mettre en évidence une modification de l’épissage du facteur de transcription RUNX1, en faveur de la forme tronquée proleucémogène RUNX1a (CALRwt vs CALRins5, Fold-increase = 3,4, p = 0,0006 ; CALRwt vs CALRdel52, Fold-increase = 3,1, p = 0,002 ; JAK2wt vs JAK2V617F, Fold-increase = 1,6, p = 0,02). Dans les cellules DM, il a été mesuré une augmentation de la production de 2-HG, reflet d’une mutation fonctionnelle d’IDH2.
Conclusion. L’expression des mutations drivers entraîne une indépendance aux cytokines, reproduisant le phénotype des SMP, ainsi qu’un changement de l’épissage de RUNX1 en favorisant la production d’un transcrit proleucémogène, par le biais de mécanismes moléculaires qui restent à élucider. D’autres anomalies de l’épissage sont recherchées par RNA-seq dans les cellules exprimant la mutation JAK2V617F seule ou en association avec une mutation épigénétique (IDH2R140Q). Les résultats de différentiels d’expression et d’épissage seront présentés.
10-17
Prise en charge thérapeutique de 342 polyglobulies de Vaquez dans les centres libéraux français
O. Fitoussi*1, M. Balhadère1, JM. Bons2, C. Lenoir1, M. Boisseau3, C. Dauriac4, AV. Doncker5, B. Bareau5, E. Legouffe6, M. Cliquennois7, S. Bologna8
1 Hématologie, Polyclinique Bordeaux Nord Aquitaine, Bordeaux ; 2 Hématologie, Centre de Radiothérapie Joseph Bellot, Montluçon ; 3 Hématologie, Clinique Pasteur, Toulouse ; 4 Oncohématologie, clinique Victor Hugo, Institut interrégional de cancérologie, Le Mans ; 5 Service d’hématologie et médecine interne, Hôpital privé Sévigné, Vivalto Santé, Cesson-Sévigné ; 6 Hématologie, Oncogard, centre hospitalier du Gard, Alès ; 7 Hématologie, Hôpital privé de Villeneuve d’Ascq - Ramsay Santé, Villeneuve-d’Ascq ; 8 Oncohématologie, Sel Du Centre D’oncologie de Gentilly, Nancy
Introduction. Les syndromes myéloprolifératifs (SMP) représentent une pathologie souvent rencontrée dans la patientèle des hématologues libéraux, réunis au sein du réseau HLA (Hématologues Libéraux Associés). Il nous est apparu intéressant de préciser, au sein de ces différents centres, quelles étaient les thérapeutiques utilisées en 2019 pour les Polyglobulies de Vaquez (PV), dans des conditions de vie réelle.
Patients et méthodes. 15 centres HLA ont enregistré, grâce à un eCRF spécifique, sur une période de six mois (du 5 novembre 2018 au 5 mai 2019), les éléments thérapeutiques de 342 patients atteints de PV. Les critères de diagnostic retenus pour les PV entre 2008 et 2016 étaient ceux de la WHO 2008 et ceux de la classification OMS 2016 pour les PV depuis 2016. Lors du recueil de données, les hématologues devaient renseigner tous les traitements (myélofrénateurs, saignées, antiagrégants, anticoagulants) utilisés depuis le diagnostic.
Résultats. L’âge moyen de la population est de 71 ans (34-99) avec 64 % d’hommes et 36 % de femmes. Tous les patients sont suivis régulièrement (fréquence moyenne : six mois) par leur hématologue et sont traités, au moment du recueil, par antiagrégants (82 %), saignées (36 %), anticoagulants (24 %), en plus du traitement myélofrénateur. Les saignées sont réalisées en moyenne cinq fois par an, pour la plupart en institution (68/338) versus 50/338 à domicile. La majorité des patients reçoivent un traitement de 1re ligne (232/298 ; 77 %) avec l’Hydroxyurée dans 91 % des cas. 69 patients sont considérés en deuxième ligne thérapeutique, 25 en troisième ligne, 11 en quatrième ligne et un patient en cinquième ligne. Le ruxolitinib est préférentiellement utilisé en deuxième et troisième lignes (43 % et 50 % respectivement), de façon inhomogène sur le territoire français. Quant à l’Interféron Pégylé, il est utilisé chez 6 % des patients en 1re ligne (20/298), 15 % en deuxième ligne (14/89) et 10 % en troisième ligne (3/28).
Discussion. L’utilisation récente des inhibiteurs des Janus kinase 1 et 2 (JAK1 et JAK2) a profondément modifié la prise en charge des PV en pratique courante dans les centres d’hématologie libérale en alternative à l’hydroxyurée lorsque celle-ci est inefficace ou par intolérance. Son utilisation est très région-dépendante dans notre étude avec une prescription plus importante dans les régions au sud de la Loire. Ces pratiques seront probablement prochainement modifiées par l’utilisation du PEG-interféron en première ligne.
Conclusion. Cette étude observationnelle menée par les centres HLA nous a permis de préciser quelle était la répartition des différentes thérapeutiques utilisées dans la prise en charge de la PV. La prise en charge est conforme aux recommandations internationales. Le Ruxolitinib semble s’imposer comme traitement de deuxième intention.
10-18
Faible incidence des mutations du gène EPOR dans les polyglobulies idiopathiques
M. Filser*1, B. Aral2, F. Airaud3, A. Chauveau4, A. Bruce5, Y. Polfrit6, A. Thiebaut-Bertrand7, M. Gauthier8, C. Le Maréchal9, E. Lippert10, S. Beziau11, C. Garrec12, B. Gardie13, F. Girodon1
1 Hématologie biologique, CHU Dijon, Dijon ; 2 Labo génétique chromosomique moléculaire, CHU DIJON, Dijon ; 3 CHU Nantes, Laboratoire de Génétique Moléculaire, Nantes ; 4 Laboratoire d’hématologie, CHRU de Brest, Brest ; 5 Hématologie pédiatrique, oncologie et soins palliatifs, University Of Alberta, Edmonton, Canada ; 6 Pédiatrie, CH Albert Bousquet, Nouméa, Nouvelle-Calédonie ; 7 Hématologie, CHU de Grenoble CHU, La Tronche ; 8 Hématologie, CHU Toulouse, Institut Universitaire du Cancer, Toulouse ; 9 Laboratoire de génétique moléculaire, CHRU de Brest, Brest ; 10 Laboratoire d’hématologie, CHU de Bordeaux, Bordeaux ; 11 Service de génétique, Hôtel-Dieu, Nantes ; 12 Génétique médicale, CHU Hôtel-Dieu, Nantes ; 13 Laboratoire de biologie et de pharmacologie appliquée (lbpa), CNRS-ENS de Cachan, LabEx LERMIT, Cachan
Introduction. Les polyglobulies sont définies par une augmentation du volume globulaire total. Les étiologies sont variées et le diagnostic est dans la majorité des cas établi assez facilement. En dehors de polyglobulies de Vaquez, les origines de l’érythrocytose restent souvent inconnues, même après des investigations approfondies, ce qui conduit au diagnostic de polyglobulie idiopathique.
Avec le développement du séquençage de nouvelle génération (NGS), les voies de signalisation impliquées dans l’érythropoïèse sont maintenant mieux décrites, permettant de mettre en évidence de nouvelles mutations et de nouveaux gènes cibles, dont celui codant le récepteur à l’érythropoïétine (EPOR).
Matériels et méthodes. La séquence du gène EPOR a été étudiée chez une large cohorte de 270 patients (229 hommes, 41 femmes) parmi un panel de gènes NGS incluant les gènes PHD1 (EGLN2), PHD2 (EGLN1), PHD3 (EGLN3), HIF-1A, HIF-2A (EPAS1), HIF-3A, VHL, VHLL, EPOR, JAK2, LNK (SH2B3), CBL et BPGM.
Tous ces patients avaient auparavant bénéficié d’explorations complémentaires afin d’éliminer toutes causes classiques de polyglobulie.
Résultats. Trois mutations différentes du gène EPOR, toutes dans l’exon 8, ont été identifiées chez trois des 270 patients testés. Deux mutations étaient déjà connues et décrites comme pathogènes (c.1142_1143delCC, p.Pro381Glnfs*2 et c.1220C>A, p.Ser407*).
Pour le troisième patient âgé de 31 ans et connu pour une polyglobulie isolée depuis l’âge de 20 ans, originaire de Nouvelle-Calédonie, la mutation retrouvée (c.1293del, p.Ser432Alafs*21) n’avait pas encore été décrite dans la littérature. La recherche de cette mutation auprès d’autres laboratoires français travaillant sur la même voie de signalisation a permis de la retrouver chez trois autres patients eux aussi originaires de Nouvelle-Calédonie mais sans lien familial connu. À noter que l’ensemble des patients avec une mutation de l’EPOR avaient des valeurs d’EPO sérique basses.
Conclusion. La découverte du même variant dans quatre familles différentes, toutes originaires de Nouvelle-Calédonie, avec une histoire familiale d’érythrocytose renforce l’hypothèse d’un impact fonctionnel de ce variant.
Cette nouvelle mutation est en effet localisée dans le domaine protéique où toutes les mutations pathogènes ont été précédemment décrites. Elle conduit à une troncature du récepteur qui devrait entraîner la perte des principaux sites de liaison des régulateurs négatifs, expliquant le phénotype observé.
Notre étude confirme que les mutations de l’EPO-R sont relativement rares chez les patients atteints de polyglobulie idiopathique (3/270), et la nécessité d’être recherchées chez les patients ayant de faibles taux sériques d’EPO et/ou des antécédents familiaux d’érythrocytose.Données hématologiques et génétiques des patients présentant une mutation d’EPOR Patient Âge Sexe GR (×1012/L) Hémoglobine (Hb, g/L) Hématocrite (Ht, %) VGM (fL) EPO (UI/L) Mutation cDNA Mutation protéine Histoire Familiale 1 52 F 5,23 172 50 1 c.1142_1143del1CC p.Pro381Glnfs*2 Père et frère 2 14 F 7,1 200 86 < 1 c.1220C>A p.Ser407* Mère et sœur 3 31 M 6,44 216 61,3 < 10,5 c.1293del p.Ser432Alafs*21 Oui 4 8 M 7,3 197 57,5 78 < 0,6 c.1293del p.Ser432Alafs*21 Oui
10-19
Les blocs intrahépatiques par thrombi plaquettaires intrasinusoïdaux pourraient expliquer l’hypertension portale des patients atteints de syndrome myélodysplasique/myéloprolifératif sans mutation
A. Normand*1, Y. Le Bris2, E. Frampas3, J. Gournay4, JF. Mosnier1
1 anatomopathologie, Hôtel-Dieu, Nantes ; 2 Laboratoire hématologie biologique, Hôtel-Dieu, Nantes ; 3 Radiologie, Hôtel dieu, Nantes ; 4 Hépato-gastro-entérologie, Hôtel dieu, Nantes
Introduction. Le but de ce travail était de corréler l’observation de la métaplasie myéloïde (ou hématopoïèse intra-hépatique) avec les lésions histologiques hépatiques associées à l’hypertension portale (HTP) et les diagnostics hématologiques de syndrome myéloprolifératif et myélodysplasique incluant la biologie moléculaire.
Patients et méthodes. Entre 2012 et 2018 des biopsies hépatiques présentant une métaplasie myéloïde (MM) ont été isolées à partir des archives du département de pathologie. Les données cliniques, hématologiques et d’imageries ont été collectées. Toutes les biopsies ont été relues et les lésions décrites dans l’HTP sans cirrhose ont été étudiées : dilatation sinusoïdale, fibrose périsinusoïdale, hyperplasie nodulaire régénérative, veinopathie obstructive portale. Une étude immunohistochimique du CD31 et la recherche des mutations JAK2/CALR/MPL ont été réalisées.
Résultats. 17 patients avec une métaplasie myéloïde ont été inclus, dont 10 patients (59 %) présentant des signes d’HTP (varices œsophagiennes, ascite, élévation du gradient porte > 5 mmHg). Dans cette sous-population, nous avons observé une dilatation sinusoïdale dans 10 cas, fibrose périsinusoïdale dans neuf cas, une hyperplasie nodulaire régénérative dans cinq cas, une veinopathie oblitérante portale dans trois cas et des thrombi plaquettaires intrasinusoïdaux CD31+ étaient observés dans trois cas.
Un syndrome myélodysplasique/myéloprolifératif (SMM) a été diagnostiqué chez six patients avec une HTP et tous montraient une fibrose périsinusoïdale, trois présentaient une thrombose porte extra-hépatique, trois une hyperplasie nodulaire régénérative, un une veinopathie oblitérante portale et deux des thrombi plaquettaires intrasinusoïdaux. Tous les patients présentant une thrombose de la veine porte extra-hépatique ou une hyperplasie nodulaire régénérative présentaient un SMM associé à la mutation JAK2V617F. Les thrombi plaquettaires intrasinusoïdaux étaient retrouvés chez un patient présentant un SMM (une leucémie myélomonocytaire chronique de type 2 et une thrombocytémie essentielle). Un de ces patients ne présentait aucune mutation JAK2/CALR/MPL. Des thrombi plaquettaires intrasinusoïdaux ont aussi été retrouvés chez un patient présentant une HTP sans SMM, ni thrombose porte extra-hépatique prouvés.
Conclusion. Bien que la plupart des HTP associées à la métaplasie myéloïde soient liées à une thrombose porte extrahépatique ou une hyperplasie nodulaire régénérative, surtout en cas de SMM avec mutation JAK2V617F, des blocs intrasinusoïdaux par thrombi plaquettaires pourraient expliquer certaines HTP, même en l’absence de mutation.
10-20
Peut-on se passer de la mesure du volume globulaire total pour affirmer une polyglobulie vraie ?
M. Grenier*1, D. Callegarin2, M. Nughe1, B. Gardie3, JM. Riedinger4, F. Girodon5
1 Hématologie Biologique, Centre hospitalier universitaire F. Mitterand Dijon-Bourgogne, Dijon ; 2 Laboratoire d’hématologie, CHU le Bocage, Dijon ; 3 Laboratoire de biologie et de pharmacologie appliquée (lbpa), CNRS-ENS de Cachan, LabEx Lermit, Cachan ; 4 Laboratoire de biologie médicale, Centre Georges François Leclerc, Dijon ; 5 Hématologie biologique, CHU Dijon, Dijon
Introduction. Une polyglobulie est habituellement associée à une augmentation de l’hémoglobine (Hb) et/ou de l’hématocrite (Ht). Cependant, pour affirmer le diagnostic de polyglobulie vraie, il est nécessaire d’obtenir la mesure du volume globulaire total (VGT) > 125 %. Cet examen est toutefois fondé sur une technique de mesure isotopique ce qui rend sa disponibilité limitée à certains centres.
L’objectif de cette étude est de déterminer s’il est possible d’affirmer une polyglobulie vraie grâce à l’utilisation d’un algorithme fondé sur les paramètres de la numération formule sanguine (NFS) examen simple, peu coûteux et accessible à tous les centres.
Matériels et méthodes. Entre 1999 et 2018, 195 mesures du VGT, associées aux paramètres de la NFS (Hb, Ht, volume globulaire moyen (VGM), nombre d’hématies, concentration corpusculaire moyenne en hémoglobine (CCMH), teneur corpusculaire moyenne en hémoglobine (TCMH) et indice de distribution des globules rouges (IDR)) ont été collectées chez des hommes (âge moyen : 56 ans, de 15 à 83 ans) avec suspicion de polyglobulie. La cohorte a été séparée en deux groupes selon le seuil de VGT de 125 %. Les moyennes de chaque paramètre érythrocytaire ont été comparées en utilisant le test statistique de Mann-Whitney (seuil 5 %). Sur la base des seuils d’Hb et d’Ht proposés par l’organisation mondiale de la santé (OMS) de 2008 et 2016, une courbe ROC a permis de déterminer la sensibilité et la spécificité de ces deux paramètres sur la cohorte étudiée.
De plus, une approche mathématique fondée sur un logiciel d’intelligence artificielle – logiciel MATLAB® – a été utilisée pour déterminer un algorithme capable de prédire le statut polyglobulique. Les sept paramètres érythrocytaires ont été considérés comme des variables prédictives numériques.
Chaque performance d’algorithme testé a été évaluée grâce aux courbes ROC et à des indicateurs incluant la précision et l’exactitude.
Résultats. 110 sujets avaient une polyglobulie vraie et 85 une fausse polyglobulie. Le paramètre érythrocytaire le plus statistiquement discriminant et le mieux corrélé avec un VGT augmenté était le nombre d’hématies (p = 0,0001), suivi par la CCMH et l’Ht (p = 0,003), l’IDR (p = 0,002) et la TCMH (p = 0,05). En revanche, aucune différence significative (p > 0,05) n’a été observée pour l’Hb et le VGM. La sensibilité et la spécificité étaient de 84 % et 22,6 % pour une Hb > 16,5 g/L, 26 % et 83 % pour une Hb > 18,5 g/L, et 89 % et 28 % pour un seuil d’Ht à 49 %. Tous les patients avec un Ht ≥ 60 % avaient une polyglobulie vraie, confirmant ce qui a déjà été rapporté dans la littérature.
Aucun des algorithmes testés par le logiciel MATLAB® n’a permis d’obtenir une exactitude supérieure à 63 % pour le diagnostic de polyglobulie vraie.
Conclusion. En dehors des cas où l’Ht est supérieur à 60 % chez les hommes, aucun des paramètres érythrocytaires, seuls ou combinés aux autres paramètres, n’est en mesure de prédire une polyglobulie vraie, et ce malgré l’utilisation d’algorithmes utilisant l’intelligence artificielle. Il reste donc nécessaire de mesurer le VGT pour confirmer l’existence d’une polyglobulie vraie.
10-21
Intérêt de l’analyse par séquençage de nouvelle génération dans les maladies de Vaquez JAK2 exon 12 positives avec une faible charge allélique
M. Filser1, B. Aral2, V. Bourgeois1, F. Airaud3, P. Martin1, C. Garrec3, S. Bezieau4, B. Gardie4, F. Girodon*1
1 Hématologie biologique, CHU Dijon, Dijon ; 2 Labo génétique chromosomique moléculaire, CHU Dijon, Dijon ; 3 Génétique médicale, CHU Hôtel-Dieu, Nantes ; 4 Génétique, Hôtel-Dieu, Nantes
Introduction. Près de 98 % des patients atteints de Polyglobulie de Vaquez (PV) sont porteurs d’une mutation JAK2, soit sur l’exon 14 (JAK2V617F) soit sur l’exon 12, dans 95 % et 3 % des cas, respectivement). Les mutations JAK2 de l’exon 12 sont variables et hétérogènes, incluant des délétions, des insertions et des duplications, c’est pourquoi les techniques basées sur la PCR allèle-spécifique (AS-PCR) sont inadaptées pour leur dépistage. D’autre part, le séquençage Sanger et le pyroséquençage n’ont pas une sensibilité suffisante pour le dépistage dans certains cas. La technique HRM quant à elle semble être une technique très efficace pour la détection des mutations de l’exon 12 de JAK2 parce qu’elle est associée à une sensibilité et une spécificité élevées ; l’analyse par HRM fournit des courbes de fusion aberrantes indiquant la présence d’une mutation qui doit ensuite être caractérisée à l’aide du séquençage Sanger. Néanmoins, des résultats faussement négatifs sont possibles avec le séquençage de Sanger même lorsqu’il est associé à des courbes de fusion aberrantes, en cas de faible charge allélique.
Patients et méthodes. Au cours des trois dernières années, 250 échantillons de patients français avec une polyglobulie idiopathique (définie par un volume globulaire total > 125 % ou un hématocrite > 60 % chez l’homme ou 56 % chez la femme) ont été testés par NGS avec un panel dédié comprenant des gènes impliqués dans la régulation des voies d’hypoxie et de l’érythropoïèse, dont le gène JAK2. En raison du coût élevé de cette technique, les patients avaient préalablement fait l’objet d’un processus de validation clinique et biologique afin d’écarter les causes évidentes de polyglobulie, en particulier, la recherche d’une mutation JAK2 sur les exons 12 et 14 devait être négative afin d’exclure une PV.
Résultats. Chez quatre patients (trois hommes et un femme, âge moyen au diagnostic 65 ans), des mutations JAK2 exon 12 à faible charge allèle ont été observées, alors qu’elles étaient initialement considérées comme négatives pour cette mutation. Les mutations observées chez ces patients étaient déjà répertoriées, mais avec une charge allélique de 6 à 13 %, inférieure au seuil de sensibilité du séquençage Sanger. À noter par ailleurs qu’aucune autre mutation (y compris LNK, EPOR, HIF, VHL ou PHD2) n’a été observée chez les quatre patients.
Une fois la présence des mutations de l’exon 12 de JAK2 détectée par l’analyse NGS, le diagnostic de PV a été confirmé et les patients ont été traités par saignée ou par cytoréducteur. En particulier, un patient avait présenté une thrombose veineuse récurrente traitée par anticoagulants oraux. Si le diagnostic de PV avait été porté plus tôt, ces épisodes thrombotiques auraient certainement pu être évités, soulignant la nécessité d’un diagnostic précis et rapide, surtout en cas de polyglobulie avec une EPO sérique basse.
Conclusion. Devant une polyglobulie à EPO basse, en l’absence d’une mutation de l’exon 12 de JAK2 par les techniques classiques, une analyse NGS est recommandée afin d’écarter une PV à faible charge allélique.
10-22
Prévalence des mutations de la calréticuline dans les néoplasies myéloprolifératives
M. Abdelghani*1, H. Hammami2, H. Ouragini3, I. Moumni2, I. Safra4, S. Menif5
1 laboratoire hématologie, institut pasteur de Tunis, Tunis, Tunisie ; 2 Laboratoire d’hématologie, Institut Pasteur de Tunis, Tunis, Tunisie ; 3 Laboratoire d’hématologie Moléculaire et Cellulaire, Institut Pasteur de Tunis, Tunis-Belvedere, Tunisie ; 4 Hématologie, Institut Pasteur, Tunis, Tunisie ; 5 laboratoire hématologie, institut pasteur, Tunis, Tunisie
Introduction. Les néoplasies myéloprolifératives (NMP) Philadelphie négatif sont des hémopathies myéloïdes clonales de la cellule souche hématopoïétique. Elles se caractérisent par une hyperplasie de la lignée myéloïde avec conservation de leur capacité de différenciation. Les NMP sont classiquement classées en polyglobulie de Vaquez (PV), thrombocythémie essentielle (TE) et myélofibrose primitive (MFP). En 2013, des mutations récurrentes dans l’exon 9 du gène de la calréticuline (CALR) ont été rapportées dans les MPN JAK2V617F négatif et incriminés dans leur physiopathologie. L’objectif de notre travail est de déterminer la prévalence des mutations CALR dans les NMP et de corréler les résultats trouvés avec les paramètres clinicobiologiques des patients.
Patients et méthodes. 115 patients suspects de NMP et négatifs pour bcr-abl et JAK2V617F ont été inclus dans l’étude. Sur l’ADN génomique extrait à partir de prélèvements de sang périphérique, une PCR utilisant des amorces flanquant l’exon 9 de calr suivie d’un séquençage bidirectionnel a permis de rechercher d’éventuelles mutations.
Résultats. Nos résultats ont conclu à une prévalence de 26 % de mutations calr dans les NMP avec une fréquence de 31 % dans les TE et 16 % dans les MFP. L’étude comparative a montré que les patients suspects de TE et mutés CALR ont un âge plus avancé et présentent un taux de plaquettes et un taux de leucocytes plus bas comparés aux TE non mutés. Les patients suspects MFP et mutés CALR présentent un taux de plaquettes, un taux de leucocytes et un taux d’hémoglobine plus élevés comparés aux MFP non mutés.
Discussion. Les mutations CALR décrites à ce jour sont localisées exclusivement au niveau de l’exon 9 et provoquent ainsi un décalage du cadre de lecture avec modification de la charge de la protéine qui active de façon constitutive la voie JAK-STAT via le récepteur MPL. Selon les données de la littérature les mutations CALR sont associées à un pronostic favorable, en effet les TE mutées thrombosent moins que les TE non mutées et les MFP mutées ont une meilleure survie globale comparées aux MFP non mutées.
Conclusion. Nos résultats sont en concordance avec les données de la littérature. Ce marqueur est un outil fiable pour la stratification des NMP.
10-23
Aspects cliniques et évolutifs de la myélofibrose primitive
KM. Benlabiod*1, N. Rekab1, F. Lamraoui1, S. Taoussi1, S. Oukid1, Y. Bouchakor Moussa1, H. Brahimi1, B. Cherifa1, C. Guezlane1, M. Mezroud2, AA. Ziani2, A. Rebouh2, MT. Abad1, M. Bradai1
1 Hématologie EHS ELCC université Blida, laboratoire de recherche sur les hémopathies malignes, et les hémoglobinopathies. Faculté de Médecine, B. P 270, Route de Soumaa, Blida, Algérie ; 2 Hématologie, EHS ELCC, Blida, Algérie
Introduction. La Myélofibrose (MF) primitive est le syndrome myéloprolifératif Philadelphie négatif (Ph1-) classique le plus rare et le plus grave ; elle est de pronostic sombre du fait de l’absence d’un traitement médical curatif ; l’allogreffe de cellules souches hématopoïétiques peut certes guérir la MF, mais l’éligibilité à cette procédure est limitée à peu de patients (pts).
Patients et méthodes. Nous rapportons l’expérience de notre centre par une étude rétrospective et descriptive incluant les pts atteints de MF primitive. Le diagnostic a été posé sur la biopsie médullaire osseuse (BMO) complété par la biologie moléculaire dans la majorité des cas.
Résultats. Notre étude porte sur vingt-sept patients (pts) : le sex-ratio est de 1,25 avec une légère prédominance masculine ; l’âge moyen est de 54 ans (24-74), Le délai diagnostique est très variable de 7 j à quatre ans. La découverte fortuite de la maladie a été notée chez trois pts à l’occasion de signes d’anémie dans quatre cas, par une douleur abdominale dans 15 cas, des signes d’HTP dans un cas, une thrombophlébite dans un cas, un syndrome de Budd-Chiari dans un cas et par une sténose de l’artère rénale chez un pt. Les signes généraux ont été notés chez 11 pts (40 %), la splénomégalie chez 25 pts (92 %), l’hépatomégalie chez trois pts (11 %). À l’hémogramme : un taux moyen d’Hb à 10 g/dL (5,2-16) ; l’anémie a été notée chez 20 pts (74 %) ; un taux moyen de GB à 15 576/mm3 (1 620-150 420) ; une leucopénie a été notée chez cinq pts (18 %) ; un taux moyen de plaquettes à 251 000 (17 000-687 000/mm3) ; la thrombopénie est notée chez sept cas (25 %). Au frottis sanguin, la présence des cellules en larmes a été notée dans 13 cas (48 %), une myélémie < 20 % dans 10 cas (37 %), les blastes circulants ont été notés dans 10 cas (37 %) avec une moyenne de 2,8 % (2-8). À la BMO le grade de la myélofibrose a été précisé dans 13 cas (48 %) dont sept cas de grade II et six cas de grade III, la mutation JAK2V617F est objectivée dans 12 cas (54 %), et la Calréticuline dans deux cas. Les patients sont répartis en groupes pronostiques selon l’IPSS (faible risque = 4 cas, intermédiaire I = 9 cas, Intermédiaire II = 9 cas, risque élevé = 5 cas) et selon le DIPSS (faible risque = 4 cas, intermédiaire I = 11 cas, intermédiaire II = 10 cas, risque élevé = 2 cas). L’hydroxyurée a été utilisée chez 16 pts (59 %), le support transfusionnel dans sept cas (25 %), une allogreffe chez un pt, et une simple surveillance dans quatre cas (14 %). Le traitement chélateur entamé chez trois pts. L’évolution a été émaillée d’une transformation blastique en LAM chez deux pts (7 %) traités par chimiothérapie classique dont un décédé en échec et un greffé après RC, une décompensation d’une HTP a été notée chez un pt. À ce jour 20 pts sont vivants en stabilité clinique et biologique, deux décédés, et cinq sont perdus de vue.
Conclusion. La myélofibrose primitive est une maladie grave dont le traitement est basé sur les scores pronostiques qui permettent d’identifier au diagnostic les patients graves, cette étude nous a permis de soulever les difficultés de la prise en charge de ces patients en absence des molécules cibles (les inhibiteurs de JAK2) qui ont clairement modifié l’histoire naturelle de la maladie en améliorant la qualité de vie des patients. L’allogreffe reste le seul traitement radical dont nous disposons mais malheureusement une minorité de patients peut en bénéficier.
10-24
Analyse des mutations JAK2V617F, CALR et MPL dans la thrombocytémie essentielle
I. Ouahchi*1, D. Hmida1, N. Bensayed2, K. Zahra2, YY. Ben2, A. Laatiri3, A. Khelif2, H. Sennana1, A. Saad1
1 cytogénétique, génétique moléculaire et biologie de la reproduction, CHU Farhat Hached, Sousse, Tunisie ; 2 Hématologie clinique, CHU Farhat Hached, Sousse, Tunisie ; 3 Hématologie clinique, CHU Fattouma Bourguiba, Monastir, Tunisie
Introduction. La thrombocytémie essentielle (TE) est un syndrome myéloprolifératif (SMP) résultant de la transformation d’une cellule souche hématopoïétique et aboutissant à la production excessive des cellules myéloïdes matures, portant essentiellement sur la lignée mégacaryocytaire. Grâce à la découverte de la mutation JAK2V617F puis de la mutation MPL exon 10 et plus récemment de la mutation CALR exon 9, les patients atteints de TE ont pu bénéficier de marqueurs moléculaires fondamentaux pour le diagnostic. L’objectif de notre étude est d’analyser la fréquence des mutations JAK2, CALR et MPL chez des patients suspects atteints de TE dans une série du centre Tunisien.
Patients et méthodes. Notre étude a été réalisée sur 52 prélèvements sanguins. La technique adoptée pour la recherche de la mutation JAK2V617F était celle de la discrimination allélique Taq man. Un kit commercial, Ipsogen JAK2 MutaScreen, contenant un mélange d’amorces et de sondes spécifiques, de l’allèle sauvage et de l’allèle muté, a été utilisé. Pour la recherche des mutations MPL et CALR, nous avons eu recours à la technique de séquençage Sanger.
Résultats. L’âge moyen des patients était de 62 ans avec une prédominance masculine. Le taux moyen de plaquettes était de 1 085 G/L. La mutation la plus fréquente était la mutation JAK2V617F, observée dans 25 (48 %) cas, suivie de la mutation CALR, détectée dans 11 (21,2 %) cas. Contrairement à la majorité des études de la littérature, le type 2 de la mutation CALR (ins TTGTC) était le plus fréquent, retrouvé chez neuf patients, suivi du type 1 (del L367FS*46) et d’un type rare (ins ATGTC), chaque type étant observé chez un patient respectivement. Enfin la mutation MPL W515L était détectée chez une seule patiente (2 %). Dans notre travail, les trois mutations JAK2, CALR et MPL étaient mutuellement exclusives. Au total, grâce à ces analyses, le diagnostic de TE a pu être confirmé chez 71,2 % des patients qui ont bénéficié ainsi d’un marqueur moléculaire, en l’occurrence l’une des trois mutations. 28,8 % des patients avaient un profil mutationnel triple négatif.
Conclusion. Nos résultats sont très encourageants pour promouvoir le diagnostic moléculaire des SMP «non LMC» en Tunisie, étant donné que la présence de l’une des trois mutations JAK2, CALR ou MPL constitue un critère diagnostique majeur du diagnostic de TE selon l’OMS 2016.
10-25
Quand l’atypie complique le diagnostic moléculaire : à propos de deux cas de syndromes myéloprolifératifs
C. Friedreich*1, L. Zalmai1, C. Bravetti1, C. Cazelles2, J. Gay1, D. Bouscary3, M. Vignon3, O. Kosmider4
1 Hématologie biologique, Hôpital Cochin, Paris ; 2 Hématologie, Hôpital Necker, Paris ; 3 Hématologie, Hôpital Cochin, Paris ; 4 Hématologie biologique, AP-HP, Hôpital Cochin, Paris
Introduction. Les syndromes myéloprolifératifs (SMP) incluant la polyglobulie de Vaquez (PV), la thrombocytémie essentielle (TE) et la myélofibrose primitive (MFP) sont caractérisés par la présence d’anomalies acquises affectant les gènes JAK2, MPL et/ou CAL-réticuline (CalR). La grande majorité de ces mutations sont récurrentes, touchent des régions dites hot-spot, et sont considérées comme causales pour le développement de la pathologie. Afin d’identifier ces mutations, les démarches des laboratoires de biologie médicale s’appuient sur des techniques standardisées et accréditées. La mutation de JAK2 identifiée dans 97 % des PV et environ 50 % des TE et MFP est la substitution c.1849G>T, aboutissant à la p.V617F. Elle est facilement identifiable et quantifiable par les méthodologies actuelles. Les mutations de CalR sont des mutations décalant le cadre de lecture dans l’exon 9 du gène. Des anomalies moléculaires plus exceptionnelles ou plus « complexes » touchant ces gènes peuvent toutefois être observées. Dans ce contexte, nous rapportons deux cas qui illustrent l’importance d’associer plusieurs techniques de biologie moléculaire afin de détecter et de caractériser correctement des anomalies moléculaires atypiques.
Résultats. La première patiente est suivie pour une TE depuis 2000, transformée en leucémie aiguë en 2018. Négatif en 2000, le bilan de SMP réalisé en 2018 met en évidence une mutation atypique de CalR affectant le domaine C-terminal, une délétion de six paires de bases observée en analyse de fragments. Pour caractériser la nature de la délétion, des analyses par séquençage Sanger et NGS faites en parallèle montrent, qu’il s’agit en réalité de deux délétions successives : une première délétion d’une paire de bases (c.1198del) suivie d’une seconde délétion de cinq paires de bases (c.1300_1304del CCCCG). Ces deux délétions aboutissent à l’apparition d’un nouveau peptide C-terminal avec remplacement des acides aminés chargés négativement par un peptide chargé positivement et à une conservation du domaine KDEL. Les mutations de CAL-R sont habituellement responsables de la perte du domaine KDEL. Ici, il y a conservation du domaine, suggérant un rôle non suffisant de la perte de ce dernier dans la physiopathologie de ce SMP. Le deuxième patient est pris en charge pour un bilan de SMP où une analyse par NGS révèle la présence d’une double anomalie au niveau du codon 617 de l’exon 14 de JAK2 : une c.1849G>T qui entraîne la mutation p.V617F et une c.1851C>T additionnelle qui n’entraîne pas de changement d’acide aminé. Les données issues du NGS nous permettent d’affirmer que ces deux mutations sont présentes sur le même allèle et à la même fréquence allélique (17 %). La recherche de la mutation p.V617F par qPCR utilisant une sonde spécifique est négative. Les mutations atypiques dans l’exon 14 de JAK2 sont rares. Elles représentent 1,8 % des PV et 1 % des SMP. Il s’agit soit de deux mutations dans le codon 617 soit d’une mutation en 617 associée à une mutation touchant un codon adjacent. Dans tous les cas, leur détection par des techniques de qPCR en discrimination allélique ou de PCR allèle spécifique ciblant la mutation c.1849G>T n’est pas possible car la mutation additionnelle empêche la fixation de la sonde ou de l’amorce spécifique.
Conclusion. Ces deux cas nous rappellent qu’en cas l’absence d’identification de mutation « récurrente » dans un SMP dont la certitude clinique ne fait aucun doute, plusieurs approches techniques sont souvent nécessaires afin d’établir de façon certaine le génotype.
10-26
Impact de la révision des critères de l’Organisation mondiale de la santé 2016 sur le diagnostic de la polyglobulie de Vaquez
KM. Benlabiod*, N. Rekab, S. Taoussi, F. Lamraoui, Y. Bouchakor Moussa, S. Oukid, MT. Abad, M. Bradai
Hématologie EHS ELCC université Blida 1, Laboratoire de recherche sur les hémopathies malignes, et les hémoglobinopathies. Faculté de médecine, BP 270, route de Soumaa, Blida, Algérie
Introduction. Les critères diagnostiques de la polyglobulie de Vaquez (PV) ont considérablement évolué depuis la première approche de classification proposée en 2001. En 2008 : cinq critères diagnostiques sont établis avec l’introduction de la mutation JAK2 découverte en 2005 (deux majeurs et trois mineurs), en 2016 : quatre critères diagnostiques sont décrits (trois majeurs et un mineur). En 2001, les seuils d’Hb retenus pour le diagnostic de PV étaient de 16,5 g/dL chez la femme et de 18,5 g/dL chez l’homme. En 2008, ces mêmes seuils ont été maintenus. En 2016 ils ont été abaissés à 16 g/dL chez la femme et à 16,5 chez l’homme. L’hématocrite est à nouveau considéré, mais à un seuil plus faible que dans les anciens critères OMS 2001 et PVSG (Polycythemia Vera Study Group).
Notre objectif est d’établir l’impact de cette révision de classification sur le diagnostic chez 22 cas de PV diagnostiqués au service d’hématologie.
Patients et méthodes. Il s’agit d’une étude prospective de 22 patients atteints de PV diagnostiqués entre 2018 et 2019. Il s’agit de 10 femmes et 12 hommes. L’âge moyen au diagnostic est de 63 ans. Pour tous les patients, une application ainsi qu’une comparaison des critères diagnostiques de la PV selon la classification OMS 2008 et OMS 2016 a été réalisée.
Résultats. Dans notre série six patients avaient une valeur d’Hb ou d’Ht inférieure au seuil défini dans la classification OMS 2008, le volume globulaire total était donc nécessaire à réaliser pour ces pts mais non fait car il n’est pas de pratique courante ; donc les 22 patients ne remplissaient pas tous ce critère diagnostique avec les deux classifications. La mutation jak2 (Exon14 et Exon12) reste un critère majeur dans les deux classifications, elle est positive chez 19 pts. En 2008, la réalisation d’une BOM n’était pas nécessaire si les critères EPO, JAK2 et hémoglobine étaient présents. En 2016, elle est devenue parmi les critères majeurs, elle nous a permis de poser le diagnostic chez les trois patients non mutés, l’érythrocytose absolue c’est-à-dire Hb > 18,5 g/dL (Ht > 55,5 %) chez l’homme et Hb > 16,5 g/dL (Ht > 49,5 %) chez la femme est ajoutée à ces trois critères pour se dispenser de la BOM.
Dans notre série, la BOM n’était pas indispensable chez 14 pts selon l’OMS 2008 vs 17 pts selon l’OMS 2016 qui avaient une érythrose absolue.
L’érythropoïétine sérique normale ou subnormale est devenue le seul critère mineur en 2016 qui a permis avec les deux premiers critères majeurs (Hb ou Ht et la BOM) de rattraper les patients non mutés, elle était basse ou normale chez tous nos patients.
Au total le diagnostic de PV a été retenu selon la classification OMS 2016 chez les 22 pts (100 %) VS 16 pts (72 %) selon la classification OMS 2008.
Conclusion. Les critères de l’OMS de 2008 étaient plus sévères que ceux de l’OMS 2016 et cela a conduit probablement à une sous-estimation potentielle du diagnostic de la PV. Dans la classification OMS 2016, la baisse des seuils de l’hémoglobine avait un impact sur le diagnostic de la PV masquée ; de plus la BOM prend toute sa place et devient un critère majeur qui peut affirmer en présence du critère mineur l’EPO un diagnostic de PV en récupérant les patients non mutés. Une nouvelle notion qui est apparue : l’érythrocytose absolue, qui permet dans certains cas, de se dispenser de la BOM au diagnostic.
10-27
Résistance à l’interféron dans une maladie de Vaquez triple positive JAK2/CALR/MPL
H. Campario*1, M. Mosca2, B. Aral3, V. Bourgeois1, P. Martin1, A. Brustel1, M. Filser1, C. Marzac4, I. Plo2, F. Girodon1
1 Hématologie Biologique, CHU Dijon, Dijon ; 2 Inserm, U1170, Université Paris-Sud, Gustave Roussy, Villejuif ; 3 Laboratoire génétique chromosomique moléculaire, CHU Dijon, Dijon ; 4 Laboratoire hématologie-immunologie, Gustave Roussy, Villejuif
Introduction. Chez les patients avec une néoplasie myéloproliférative (NMP), l’interféron pégylé-α (IFN) est un traitement très efficace qui entraîne des réponses cliniques, hématologiques, moléculaires et histopathologiques.
Résultats. Un homme de 60 ans avec une PV mutée JAK2V617F avec une fréquence allélique (FA) de 30 % a été traité par HU pendant quatre ans entraînant une RHC avec 14 g d’HU/semaine et une diminution de la FA de JAK2V617F de 30 % à 3 %. En raison d’une légère intolérance à l’HU et d’un âge relativement jeune, l’HU a été progressivement remplacée par l’IFN. Néanmoins, deux mois après l’arrêt de l’HU, une perte de la RHC a été observée et la numération plaquettaire ainsi que l’hématocrite ont augmenté. L’IFN a alors été augmenté à 180 μg/semaine, mais aucune RHC n’a été obtenue. Des doses modérées d’HU (9 g/semaine) ont donc été réintroduites pendant une période de huit mois. Une RHC et une bonne RMC ont été obtenues puisque la FA de JAK2V617F dans les granulocytes périphériques est restée stable à 3 %. Enfin, après 17 mois de traitement combinant de faibles doses d’HU et 180 μg/semaine d’IFN, le traitement à l’IFN a été jugé inefficace et arrêté. Une analyse de séquençage de nouvelle génération a montré au moment du diagnostic une mutation JAK2V617F isolée, alors qu’après quatre ans de traitement à l’HU puis 17 mois de traitement combiné HU/IFN, ont été notées non seulement la mutation JAK2V617F mais aussi une mutation CALRdel52 ainsi qu’une mutation MPLS505N. Respectivement, la FA des mutations JAK2V617F, CALR et MPL était de 2 %, 37 % et 38 % après quatre ans de traitement de l’HU avant l’IFN, 3 %, 35 %, 56 % à la fin du traitement de l’IFN et à 10 %, 37 %, 47 % quatre mois après l’arrêt du traitement. Dans le même temps, l’analyse de l’ADN des follicules pileux n’a montré aucune mutation, confirmant le caractère somatique des mutations JAK2, CALR et MPL. Nous avons pu observer l’architecture clonale de l’hématopoïèse en génotypant les colonies de progéniteurs à différents temps du traitement. Tout d’abord, JAK2V617F et CALR del52 ont été trouvés dans deux clones indépendants, confirmant la nature mutuellement exclusive des mutations CALR et JAK2. Au moment du traitement par IFN, les progéniteurs homozygotes JAK2V617F ont été détectés à une très faible fréquence (1 % des progéniteurs engagés) par rapport aux progéniteurs hétérozygotes CALR del52 (50 % des progéniteurs engagés). Nous avons observé une légère augmentation temporaire des progéniteurs JAK2V617F suivie d’une réponse moléculaire complète à M15 puis d’une rechute à M18. En revanche, les progéniteurs CALR del52 ont augmenté de façon continue sous l’IFN. De plus, nous avons génotypé chaque progéniteur pour la mutation MPLS505N par HRM au début et à la fin du traitement à l’IFN. MPLS505N est apparu dans un progéniteur CALR del52 De plus, nous avons observé une expansion du clone CALR del52/MPLS505N au cours du traitement de 70 % à 86 % des progéniteurs mutés CALR.
Conclusion. Ce patient très atypique avec des anomalies génotypiques complexes nous a permis de clarifier l’impact du traitement sur les mutations : nos données mettent en évidence l’acquisition des mutations CALRdel52 et MPLS505N sous traitement HU et l’inefficacité de l’IFN sur les clones mutés CALR et CALR/MPL par rapport aux cellules de clones mutés JAK2V617F. Nos résultats soulignent l’intérêt d’une analyse NGS avant d’initier un traitement à l’IFN, ainsi que dans les cas de NMP résistants à l’IFN.
10-28
Apport de la biopsie ostéomédullaire chez les patients ayant un syndrome de Budd-Chiari
L. Chtourou*1, H. Smaoui1, M. Ghorbel2, M. Mdhaffar2, M. Boudabbous1, H. Gdoura1, M. Elloumi2, A. Amouri1, L. Mnif1, N. Tahri1
1 Service de gastro-entérologie, CHU Hédi Chaker, Sfax, Tunisie ; 2 Service d’hématologie, CHU Hédi Chaker, Sfax, Tunisie
Introduction. Le syndrome de Budd-Chiari (SBC) primitif complique un état d’hypercoagulabilité. Les syndromes myéloprolifératifs représentent la cause la plus fréquente. Le but de ce travail est de préciser l’apport de la biopsie ostéomédullaire chez les patients ayant un SBC et d’étudier les particularités cliniques et thérapeutiques de ces patients.
Patients et méthodes. Dans une étude rétrospective nous avons colligé tous les patients ayant un SBC sur une période entre janvier 2014 et avril 2018. Le diagnostic de SBC a été confirmé par une imagerie comportant une échographie doppler et/ou un angioscanner hépatique. Une biopsie ostéomédullaire a été pratiquée pour tous les patients. Pour chaque patient, nous avons recueilli les données épidémiocliniques, paracliniques et thérapeutiques.
Résultats. Au total, 12 patients étaient colligés dont sept hommes et cinq femmes. L’âge moyen était de 49 ans. Le mode d’installation était aigu dans 33,3 % des cas. Les symptômes cliniques révélateurs étaient dominés par la douleur abdominale (83 %). À l’examen, une circulation veineuse collatérale et une ascite étaient notées chez 91 % des patients, une hépatomégalie chez sept patients et une splénomégalie chez sept patients. À l’endoscopie digestive haute, des varices œsophagiennes étaient objectivées chez 83 % des patients. La thrombose avait intéressé les trois veines sus-hépatiques chez 11 patients avec une atteinte associée de la veine cave inférieure chez 27 % des patients et une thrombose de la veine porte chez deux patients. La biopsie ostéomédullaire était contributive au diagnostic chez cinq patients concluant à des syndromes myéloprolifératifs dont un cas de polyglobulie de Vaquez, trois cas de thrombocytémie essentielle et un cas de myélofibrose. Il s’agissait de trois femmes et deux hommes d’âge moyen de 49,5 ans. La thrombose était étendue à la veine cave inférieure chez trois patients et au tronc splénomésaraïque chez un patient. Dix patients étaient mis sous traitement anticoagulant dont trois étaient perdus de vue. Une reperméabilisation des veines était notée chez un patient et une extension thrombotique chez deux patients. Une complication hémorragique était notée chez quatre patients. Parmi les patients qui avaient un syndrome myéloprolifératif, un patient était perdu de vue, un patient était décédé par extension thrombotique et chez les trois autres il y avait une persistance de la thrombose sans extension.
Conclusion. Le SBC est une affection rare dont les causes sont dominées par les états prothrombotiques acquis notamment les syndromes myéloprolifératifs. La prise en charge de ces patients présente un challenge entre un traitement anticoagulant et un risque hémorragique lié à l’hypertension portale.
10-29
Myélofibrose primitive : étude monocentrique à propos de 38 cas
I. Ben Amor*1, A. Koubaa1, F. Bensaid2, I. Frikha1, M. Charfi1, R. Kallel3, M. Ghorbel1, F. Kallel1, O. Kassar1, M. Mdhaffar1, S. Hdiji1, M. Elloumi1
1 Hématologie clinique, CHU Hédi Chaker, Sfax, Tunisie ; 2 Hématologie clinique, Hôpital régional Gabés, Gabès, Tunisie ; 3 Laboratoire d’anatomopathologie, CHU Habib Bourguiba, Sfax, Tunisie
Introduction. La myélofibrose primitive (MFP) est un syndrome myéloprolifératif chronique caractérisé par une myélofibrose évolutive et une métaplasie hépatosplénique. Son pronostic est globalement péjoratif et son traitement est essentiellement symptomatique. Dans ce travail nous présentons les caractéristiques diagnostiques, pronostiques et thérapeutiques des patients suivis pour MFP au service d’hématologie de Sfax.
Patients et méthodes. Il s’agit d’une étude rétrospective ayant concerné les patients atteints d’une myélofibrose primitive diagnostiqués et pris en charge au service d’hématologie sur une période de 13 ans entre 2006 et 2018.
Les données épidémiologiques, diagnostiques et évolutives ont été recueillies.
Le diagnostic de MFP s’est basé sur les critères diagnostiques de l’OMS 2008.
Le score pronostic DIPSS a été adopté pour la stratification pronostique.
La survie globale a été calculée selon la méthode de Kaplan Meyer.
Résultats. Nous avons colligé 38 patients, 17 hommes et 21 femmes avec un sex-ratio de 0,8.
L’âge moyen était de 59 ans (24-88). Le syndrome anémique a été la circonstance de découverte la plus fréquente (52 % des cas) suivie par la pesanteur de l’hypocondre gauche (25 %). L’anémie a été constante avec un taux moyen d’hémoglobine à 7,8 g/dL. Une hyperleucocytose et une thrombocytose ont été observées chez respectivement 47 % et 25 % des patients. L’étude cytogénétique a été contributive dans 24 cas dont neuf cas (38 %) ont révélé des anomalies cytogénétiques. La recherche de la mutation jack 2 a été positive dans 55 % des cas avec un cas de mutation W515L positive. La répartition de nos patients selon le score DIPSS a été comme suit : risque faible : 10 %, risque intermédiaire 1 : 29 % et intermédiaire 2 : 50 %, haut risque : 10 %. Le support transfusionnel par des culots globulaires était indiqué chez 60 % des patients. Le traitement par Hydréa seul ou associé à une corticothérapie a concerné 50 % des patients. Les androgènes ont été prescrits en deuxième ligne chez quatre patients permettant l’obtention d’une réponse sur l’anémie chez deux d’entre eux.
Dans le cadre d’un essai clinique mené entre 2013 et 2016, cinq patients ont été traités par les anti-jak2 avec une réponse constante mais partielle sur la splénomégalie et les signes généraux avec une perte de cette réponse après l’arrêt du traitement.
Une transformation en leucémie aiguë a concerné quatre patients dans un délai respectif de 6,7,18 et 60 mois. Les complications infectieuses ont concerné quatre patients neutropéniques avec un choc septique aboutissant au décès chez deux patients. L’évolution vers un tableau d’hypertension portale a concerné 10 patients dont un a été associé à un syndrome de Budd-Chiari. Une hémochromatose a compliqué l’évolution de cinq patients. Sept patients ont été perdus de vue. La SG à cinq ans était de 39 %.
Conclusion. Notre série est caractérisée par une prédominance féminine, un âge jeune et la profondeur des cytopénies. Cependant, la fréquence des anomalies cytogénétique et la positivité de la mutation jak2 sont comparable aux données de la littérature.
La survie et la qualité de vie de nos patients sont insuffisantes incitant à délivrer des thérapeutiques plus efficaces (allogreffe, les anti-jak2).
10-30
Double mutation JAK2L611V/V617F dans un cas pédiatrique de polyglobulie
B. Lebecque1, V. Greze2, T. Tassin3, R. Veyrat-Masson3, G. Mareynat4, N. Boiret-Dupré4, N. Tribalat3, M. Berger5, C. Bourgne*6
1 Laboratoire d’hématologie, CHU Estaing, Clermont-Ferrand, EA 7453 CHELTER, Université Clermont Auvergne ; 2 Oncohématologie pédiatrique, CHU Clermont-Ferrand, Clermont-Ferrand ; 3 Hématologie biologique, CHU Estaing, Clermont-Ferrand ; 4 Hématologie biologique, CHU Estaing, Clermont-Ferrand, France, Clermont-Ferrand ; 5 Hématologie biologique, CHU Estaing, Clermont-Ferrand ; 6 Hématologie biologique, CHU Clermont-Ferrand, Clermont-Ferrand
Introduction. La Polyglobulie de Vaquez (PGV) est un syndrome myéloprolifératif (SMP) connu chez les adultes d’âge > 50 ans, caractérisé par la présence d’une masse sanguine élevée et dune mutation ponctuelle JAK2V617F (95 % des cas). Seuls 0,1 % des patients atteints de PGV ont moins de 20 ans, et leur profil biologique reste mal décrit. En particulier, l’incidence de la mutation JAK2V617F est variable d’une étude à l’autre. Nous rapportons ici le cas d’une enfant de cinq ans avec une polyglobulie associée à une double mutation JAK2L611V/V617F.
Résultats. Cette enfant a été hospitalisée suite à un épisode de fièvre et un prurit. Le bilan sanguin a montré à la fois une polyglobulie (hémoglobine 18,8 g/dL, hématocrite 56,7 %) et une augmentation du taux d’éosinophiles (3,8 × 109/L). La numération plaquettaire était normale (223 × 109/L). L’analyse cytologique de la moelle osseuse, la cytogénétique et l’immunophénotypage étaient sans anomalie.
Nous avons retrouvé une mutation JAK2V617F positive à 3 % (Ipsogen JAK2 MutaQuant Kit, Qiagen) et un taux d’érythropoïétine plasmatique abaissé (< 1,6 mUI/mL). Aucune mutation n’a été trouvée dans l’exon 12 du gène JAK2. L’analyse NGS (MYS solution, SOPHiA GENETICS, Saint Sulpice, Suisse) a révélé la présence de deux variants dans l’exon 14 du gène JAK2. La première confirmait la mutation classique JAK2V617F (c.1849G>T ; p.Val617Phe ; COSV67569051) et la seconde était en position 1832 de l’ADNc (c.1832T>C ; p.Leu611Ser ; COSV67633764). Les deux variants présentaient une VAF de 24 %, suggérant une origine somatique et la présence d’un seul clone. La visualisation IGV (Integrative Genomics Viewer) a montré que les deux variants étaient situés sur les mêmes reads. La différence entre les VAF obtenues en qPCR et NGS pourrait s’expliquer par le fait que la deuxième mutation (c.1832T>C ; p.Leu611Ser) perturbe l’hybridation entre l’amorce sens et l’ADN cible, entraînant une amplification limitée de l’ADN muté.
Conclusion. Le diagnostic de Polyglobulie primitive de type maladie de Vaquez a été retenu et un traitement par Hydroxyurée a été recommandé à 20 mg/kg/jour pour contrôler le nombre de globules rouges. À notre connaissance, il s’agit de la première description d’une double mutation JAK2L611V/V617F chez un enfant atteint de PGV. Cette mutation JAK2L611S a été rapportée seule chez deux enfants : le premier porteur d’une mutation constitutionnelle associée à un phénotype de thrombocytose (Aral et al., Haematologica 2018) et le second atteint de leucémie lymphoblastique aiguë (Kratz et al., leukemia 2006). La coexistence des deux variants L611V/V617F a été rapportée quatre fois chez des adultes présentant un phénotype de polyglobulie (Nussenzveig et al., Leukemia & Lymphoma 2012 ; Cleyrat et al., Leukemia 2010). La présence de cette double mutation L611V/V617F augmenterait l’activation de JAK2, AKT et ERK1/2 (et non STAT5) et serait associée à une érythrocytose isolée (Cleyrat et al., Leukemia 2010).
10-31
Profil épidémiologique, clinique, thérapeutique et évolutif des syndromes myéloprolifératifs type classique
FE. Rida*, M. Qachouh, S. Berrada, N. Khoubila, S. Cherkaoui, M. Lamchahab, M. Rachid, M. Abdellah, A. Quessar
Hématologie clinique et oncologie pédiatrique, Hôpital 20-Août, Casablanca, Maroc
Introduction. Les syndromes myéloprolifératifs Philadelphie négatif (polyglobulie de Vaquez, thrombocytémie essentielle et myélofibrose primitive) représentent un groupe de pathologies secondaire à une myéloprolifération clonale induite par trois mutations pilotes que sont le JAK2, la CALR et le MPL. Ces dernières ont une fréquence de distribution respective de 99, 0 et 0 % pour PV ; 55 ; 22 et 3 % pour la TE et 65 %, 20 % et 7 % pour MFP.
L’objectif de notre travail est de définir les caractéristiques clinicobiologiques, thérapeutiques et évolutives des syndromes myéloprolifératifs Philadelphie négatif.
Patients et méthodes. Étude rétrospective monocentrique, sur une période de six ans allant de 2012 à 2017. Les données ont été recueillies à partir des dossiers médicaux. La méthodologie statistique a été basée sur le calcul des moyennes de chaque variable étudiée qui nous ont permis de décrire les cas. Les variables étudiées concernent les caractéristiques épidémiologiques des patients (âge, sexe, provenance, couverture sociale…), Clinique (motif de consultation, examen clinique), biologiques (la NFS, BOM, caryotype, biologie moléculaire, etc.) et thérapeutique (lignes thérapeutiques et réponses). L’analyse de données a été faite par logiciel Excel.
Résultats. Ont été colligés 70 patients. L’âge médian était de 62 ans avec un sex-ratio à 1. 43 % avaient une thrombocytémie essentielle, 34 % une polyglobulie de Vaquez et 24 % une myélofibrose primitive. Le mode de découverte était fortuit dans 16 % des cas. La survenue d’événements thromboemboliques avant le diagnostic a été rapportée dans 12 % des cas. Sur le plan clinique une splénomégalie était présente chez 28 % des patients. La mutation JAK2V617F a été recherchée chez 72 % des patients et est revenue positive chez 53 %. La mutation de l’exon 12 de JAK2 a été recherchée chez un seul patient revenue négative. La mutation du gène CARL a été retrouvé chez un patient e tla mutation MPL n’a été recherchée chez aucun patient. Sur le plan thérapeutique, l’antiagrégant plaquettaire a été utilisé dans 52 % des cas. Le traitement cytoréducteur utilisé était l’hydroxyurée (80 % des cas) avec une réponse complète ou partielle chez 52 % des patientes et 48 % étaient résistants ou intolérants au traitement. Une deuxième ligne thérapeutique a été proposée chez deux patients : l’interféron chez une femme enceinte avec une bonne réponse et le Ruxolitinib chez le deuxième patient avec un échec thérapeutique. Sur une médiane de follow-up de 60 mois, nous retrouvons 10 % (n = 7) de transformation en LAM et une survie globale de 52 %.
Conclusion. L’étude nous a permis de faire un état des lieux et des pratiques cliniques concernant le diagnostic, le traitement et l’évolution des SMP non-Phi. Cependant une étude multicentrique avec une accessibilité d’un panel biologique pour le diagnostic pourrait permettre une meilleure étude du profil de nos patients et évaluer les résultats thérapeutiques.
 This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License
This work is licensed under a
Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License