Epileptic Disorders
MENUTwo mutations in the nicotinic acetylcholine receptor subunit A4 (CHRNA4) in a family with autosomal dominant sleep-related hypermotor epilepsy Volume 22, issue 1, February 2020

Figures
Autosomal dominant frontal lobe epilepsy with sleep-related seizures (sleep-related hypermotor epilepsy; ADSHE) is a focal epilepsy with predominantly sleep-related seizures of mostly frontal semiology. Several genes have been linked to ADSHE or autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), as it used to be called: the genes encode for several different subunits of the nicotinic acetylcholine receptor (CHRNA4, CHRNA2, and CHRNB2) (Steinlein et al., 1995; Fusco et al., 2000; Aridon et al., 2006; Becchetti et al., 2015), a potassium channel (KCNT1) (Lim et al., 2016), corticotropin releasing hormone (CRH) (Sansoni et al., 2013) and DEP domain-containing protein 5 (DEDPC5) (Picard et al., 2014). However, only approximately 20% of ADSHE patients with a positive family history carry a known mutation -and even less of the sporadic cases- indicating that not all disease-causing genes and mutations have been identified yet (Kurahashi and Hirose, 2018). Here, we present a family with ADSHE carrying two mutations, of which one is novel, on the same allele of the CHRNA4 gene.
Case study
The 23-year-old female index patient was the first in her family to seek medical counselling for seizures that started at the age of 10. The seizures mostly began with a slight movement of the head, pursing of the lips (“chapeau-de-gendarme-sign”), heavy breathing and a short-lasting bilateral tonic posture, sometimes followed by tonic elevation or small movements of the right arm. Seizure duration was between 10 and 40 seconds with immediate regain of consciousness on testing. All events were sleep-related.
Additionally, our patient reported infrequent attacks of a different semiology in which she got up from bed, spoke, turned on the light, opened the door or performed complex motor actions. These occurred more often after alcohol consumption and she sometimes recollected a dream when awakening. We did not record any of these attacks. The complex and variable nature of their semiology suggests a different entity. These attacks most likely represent NREM parasomnia.
The patient was born at term to non-consanguineous parents. During her first hour of life, she developed cyanosis and was kept in neonatal care for four weeks. A specific diagnosis of this condition was not established. She did not experience any further attacks of cyanosis, and her childhood development was normal. Comorbidities included recurring migraine with visual aura, psoriasis with arthritis and mild maternally inherited restless legs syndrome. She experienced rare sleep paralyses and hypnopompic hallucinations. She was not on any regular medication.
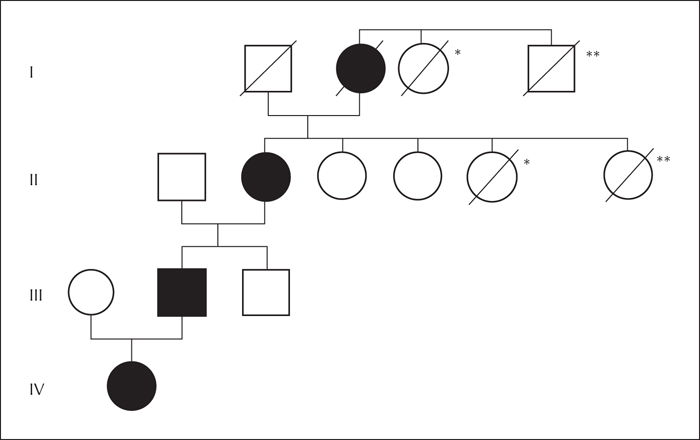
Seizure onset in her father (56 years old) occurred at the age of two, with a very similar semiology: the family described that he pursed his lips with open eyes, then held his breath and repeatedly lashed out with his arms and legs for 10 to 30 seconds. When woken up, he promptly regained consciousness. The index patient's paternal grandmother (79 years old) had seizures of an identical semiology since her adolescence. Additionally, she could describe the same seizures in her own mother, our index patient's great-grandmother. She had observed these seizures regularly during World War II when her mother was sleeping in the kitchen during the day, after keeping watch of her family during bombing attacks at night. For a family pedigree, see figure 1.
The index patient's neurological examination and cranial MRI were normal. Interictal video-EEG was normal. We recorded more than 100 seizures exclusively arising during NREM-sleep, mostly during sleep stage N2, and rarely N3. At clinical seizure onset, we recorded a relative electrodecrement followed by rapid bifrontal paroxysmal fast activity of 13-16 Hz with an evolution to rhythmic bifrontal theta activity and a mild rise in heart rate. Ictal EEG did not allow lateralization.
The case and family history together with the video-EEG monitoring led to the diagnosis of ADSHE. Genetic testing revealed heterozygosity in all three patients for two missense variants on the same allele of the CHRNA4 gene: c.742A>T (Ile248Phe) and c.1708G>C (Gly570Arg); numbers are in accordance with reference NP_000735. Neither variant has previously been published. The c.742A>T variant has not been detected in database resources in controls and the c.1708G>C variant was identified five times (gnomad; http://gnomad.broadinstitute.org/variant/20-61981055-C-G). Most software tools for the prediction of pathogenicity classified the c.1708G>C variant as probably pathogenic (fathmm-mkl, LRT, MutationTaster, PROVEAN, SIFT, MutationAssessor) with only one exception (fathmm), whereas the c.742A>T variant was classified as probably pathogenic by some (fathmm-mkl, LRT, MutationTaster, PROVEAN, SIFT) and as benign by others (fathmm, MutationAssessor). No other variants in the CHRNA4 or CHRNA2 genes were detected. Whole-exome sequencing was not performed.
Discussion
The family is affected by frontal lobe epilepsy with sleep-related seizures (sleep-related hypermotor epilepsy). Sequencing of the CHRNA4 and CHRNA2 genes did not reveal any known mutation, but two new variants in the CHRNA4 gene. Whole-exome sequencing was not performed which may be a minor limitation to our study, as we cannot exclude additional mutations in genes other than CHRNA2 and CHRNA4. However, there are a number of arguments supporting that the two variants are indeed pathogenic. First, they are located in highly conserved regions of exons 5 and 6 of a gene with known disease-causing mutations. Secondly, we detected the same variants in cis configuration (on one allele) in all three living affected family members. Our patient has no siblings, so unfortunately, we were not able to obtain DNA samples from unaffected family members.
Although we find it plausible that our CHRNA4 variant(s) are pathogenic, we cannot determine whether one or the other of the variants alone or the combination of both are pathogenic. Software tools for the prediction of pathogenicity might favour the c.1708G>C variant, however, this variant -in contrast to the second- was found five times in a population of healthy controls, which could argue against its pathogenicity, at least as a single variant. Considering the possibility of reduced penetrance which is frequent in ADSHE (Kurahashi and Hirose, 2018), there is no clear evidence that points to either of the two variants as pathogenic on their own.
Unfortunately, we could not record any of our patient's reported attacks with less stereotypical semiology. Considering the case history, we judged them most likely to represent NREM parasomnia. Nocturnal frontal lobe epilepsy patients frequently exhibit both frontal lobe seizures and parasomnia (Bisulli et al., 2010). This may be due to a shared pathophysiology for the two conditions involving the cholinergic arousal system and frequent sleep stage shifts due to the fact that seizures may be additional triggers for parasomnic events (Halász et al., 2012).
Disclosures
S. Biskup and B. Dräger have no competing interests. L. Langenbruch has received honoraria for lecturing from Eisai. G. Möddel has received honoraria for lecturing and travel expenses from UCB Pharma, Eisai, Desitin, and Electrical Geodesics Inc. (EGI). P. Young has received honoraria for lecturing and advisory boards of Löwenstein Medical, Sanofi Genzyme, Biogen, VANDA, Medice, UCB Pharma, and Biogen.