Epileptic Disorders
MENUSialidoses Volume 18, supplement 2, September 2016

Figures
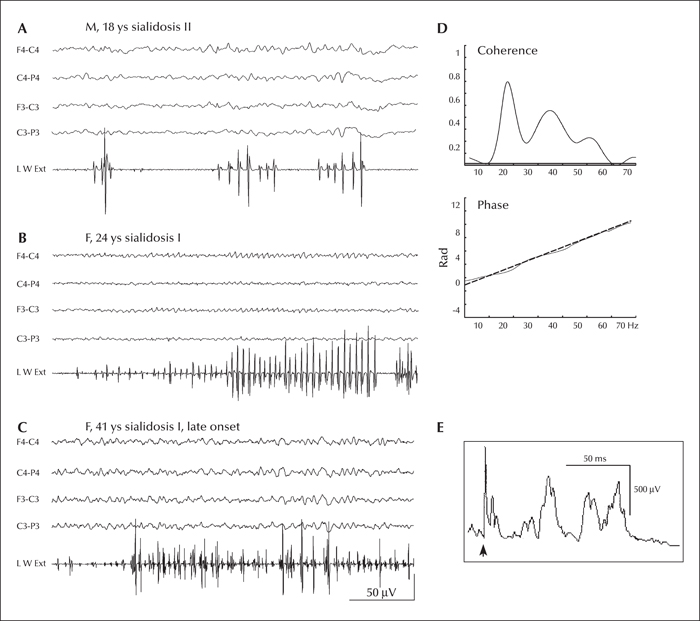
- Key words: cortical myoclonus, neuraminidase, NEU1, cortico-muscolar coherence, progressive myoclonus epilepsies
- DOI : 10.1684/epd.2016.0845
- Page(s) : 89-93
- Published in: 2016
Sialidoses are autosomal recessive disorders caused by NEU1 gene mutations and are classified on the basis of their phenotype and onset age. Sialidosis type II, with infantile onset, has a more severe phenotype characterized by coarse facial features, hepatomegaly, dysostosis multiplex, and developmental delay while patients with the late and milder type, known as “cherry red spot-myoclonus syndrome” develop myoclonic epilepsy, visual impairment and ataxia in the second or third decade of life. The diagnosis is usually suggested by increased urinary bound sialic acid excretion. We recently described genetically diagnosed patients with a specially mild phenotype, no retinal abnormalities and normal urinary sialic acid. This observation suggests that genetic analysis or the demonstration of the neuraminidase enzyme deficiency in cultured fibroblasts are needed to detect and diagnose mildest phenotypes.