Epileptic Disorders
MENUNeuronal ceroid lipofuscinoses Volume 18, supplement 2, September 2016



The neuronal ceroid lipofuscinoses (NCLs) represent a heterogeneous group of genetically-determined neurodegenerative conditions that are characterized by a progressive decline of cognitive and motor capacities, retinopathy evolving into blindness, variable cerebellar atrophy, and myoclonic epilepsy, leading to significantly decreased life expectancy (Mitchison et al., 1998; Santavuori et al., 2000; Jalanko & Braulke, 2009; Warrier et al., 2013). They are the most prevalent neurodegenerative disorders of childhood with an incidence in the USA estimated at 1.6-2.4/100,000, while in Scandinavian countries the incidence varies between 2-2.5/100,000 in Denmark, 2.2/100,000 in Sweden, 3.9/100,000 in Norway, 4.8/100,000 in Finland, and 7/100,000 in Iceland (Uvebrant & Hagberg, 1997; also reviewed in Mole et al., 2011).
The first description in the medical literature was probably by Otto Christian Stengel (1795-1890), a German physician who served in the mining community of Røros, Norway, between 1821 and 1882. He described a juvenile-onset disorder with blindness and progressive dementia (Stengel, 1826). In 1903, Frederick Eustace Batten (1865-1918), an English neurologist and paediatrician, described a similar clinical disorder and was the first to describe the neuropathology of cerebral degeneration with ocular macular changes in two members of a family (Batten, 1903). In 1905, Walther Spielmeyer (1879-1935), who had succeeded Aloïs Alzheimer as director of neurology and clinical psychiatry laboratory in Munich and Heinrich Vogt (1875-1936) reported a similar disorder (Spielmeyer, 1905; Vogt, 1905). At this time, juvenile neuronal ceroid lipofuscinosis was referred to as Batten-Spielmeyer-Vogt disease. Later, Jan Janský (1873-1921) and Max Bielschowsky (1869-1940) described a similar disorder, but with a “late-infantile” onset (Janský, 1908; Bielschowsky, 1913). This form came to be known as “Janský-Bielschowsky disease” or “late-infantile NCL”.
Hugo Kufs (1871-1955), from Leipzig, Germany, published four reports between 1925 and 1931 in which he described an adult-onset disease with similar pathological characteristics, but without the loss of vision that was so prominent in juvenile NCL and late-infantile NCL (Kufs, 1925). This came to be known as adult-onset NCL or Kufs disease. More recently, Matti Haltia (b. 1939) and Pirkko Santavuori (1933-2004), while investigating a child suspected to have GM1 gangliosidosis type II, concluded by identifying a novel type of NCL with early onset (Haltia et al., 1973a; Haltia et al., 1973b; Santavuori et al., 1973). Classic infantile NCL is also known as Haltia-Santavuori disease.
Traditionally, NCLs were classified according to the age at onset as: infantile (INCL), late-infantile (LINCL), juvenile (JNCL) and adult (ANCL), but were also known by their eponyms Haltia-Santavuori disease, Jansky-Bielschowsky disease, Batten-Spielmeyer-Vogt disease, and Kufs disease, respectively (table 1 and figure 1). Moreover, the term ‘Batten disease’ was used in the literature to designate both the whole group of NCL and JNCL in particular. As less common forms of NCL began to be discovered, these were often referred to by the country of origin of the first described patients (table 1).
Today, at least 14 affected genes are implicated, from CLN1 to CLN14, 13 of which have been identified (CLN1-8 and CLN10-14). CLN9 refers to the predicted locus in a family who do not appear to have mutations in any of the known genetic forms (Schulz et al., 2004). The currently genetically identified types of NCL are listed in table 2.
Very recently, a new nomenclature has been discussed internationally and subsequently proposed which is gene-based and specific to phenotypic variation arising from different mutations. This is an axial diagnostic classification system that includes seven axes:
- –affected gene (CLN gene symbol);
- –mutation diagnosis;
- –biochemical phenotype;
- –clinical phenotype;
- –ultrastructural features;
- –level of functional impairment; and
- –other remarks (additional genetic, environmental, or clinical features) (Mole & Williams, 2013).
In reality, NCL classed according to the affected gene, combined with the age at onset, is sufficient for general use (e.g. classic infantile CLN1 disease, or adult CLN1 disease).
Pathophysiology of NCLs
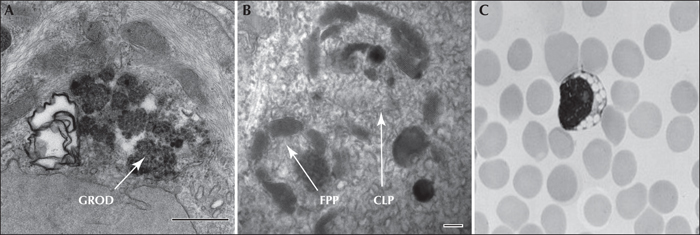
NCL are grouped together on pathological grounds due to the common presence of neuronal and extraneural autofluorescent pigment accumulations, despite diverse underlying biochemical aetiologies. They are considered lysosomal storage diseases, however, NCLs also exhibit characteristics that distinguish them from lysosomal storage diseases (Mink, 2010). Consistent with lysosomal storage disorders (LSD), many of the identified NCL proteins are present in the lysosomes, and lipofuscin-like ceroid lipopigments also accumulate in the lysosomes. Under the electron microscope, the accumulated material takes different forms: granular osmiophilic deposits (GRODs), curvilinear profiles (CLP), fingerprint profiles (FPP), as well as rectilinear complex (RLC) or so called ‘condensed forms’. However, unlike classic lysosomal storage disorders, for the NCL, the ‘stored’ material is not disease-specific. While the major clinical NCL subtypes (CLN1, CLN2 and CLN3 diseases) were formerly each associated with characteristic inclusions under electron microscopy (GRODs, CLP, and FPP, respectively) (figure 2), the ultrastructural findings do not absolutely correlate with clinical presentation, and the same NCL may contain more than one pattern of inclusion (table 3). Furthermore, the appearance of the pathological inclusions can depend on the tissue examined. Vacuolated lymphocytes are typically seen in classic juvenile CLN3 disease.
CLN1
CLN1: Genetics
In CLN1 disease, the underlying defect is the lack of activity of the lysosomal palmitoyl protein thioesterase (PPT1), an enzyme that removes palmitate residues from proteins (Vesa et al., 1995). The main protein component of the storage material is saposins A and D, with a characteristic ultrastructure of GRODs. To date, 64 mutations have been described in CLN1 (for updated information on NCL mutations see the online database at http://www.ucl.ac.uk/ncl/). The exact physiological function and in vivo substrates of PPT1 are unknown, but it is proposed that PPT1 is required to maintain various cellular processes, including apoptosis, endocytosis, vesicular trafficking, synaptic function, and intracellular signalling (Greaves & Chamberlain, 2007). In neurons, PPT1 is found also outside the lysosomal compartment in presynaptic terminals (Ahtiainen et al., 2006), suggesting that PPT1 is not exclusively confined to lysosomes, and the disease is not due solely to the abnormal storage.
CLN1: Clinical presentation
The first symptoms of classic infantile CLN1 disease manifest in the second half of the first year of life, with irritability, followed by rapid psychomotor deterioration, central hypotonia, and deceleration of head growth. These are quickly followed by myoclonic jerks (and other seizures types) and blindness with optic atrophy. The ERG (electroretinogram) is unrecordable by age 4 years. Hand-wringing often develops during the disease course, which, along with the slowing of head growth and developmental regression, raises the differential for Rett syndrome, but unlike the latter, CLN1 disease does not stabilize, continuing instead to deteriorate until death in early childhood (Mole et al., 2005). Most children die at around 10 years of age.
Of all the NCLs, CLN1 disease has the widest range of age at onset, determined by the combination of particular mutations. Although the majority of patients have infantile onset, some have late-infantile, juvenile, and even adult-onset, as late as 40 years of age (Van Diggelen et al., 2001, Ramadan et al., 2007).
CLN1: Diagnosis
Historically, the diagnosis was made based on the ultrastructural finding of GRODs, together with the presence of suggestive clinical features. The diagnosis can now be made rapidly and specifically by demonstrating a lack of PPT1 activity, even in adult-onset forms.
Brain MRI usually demonstrates a variable degree of cerebral atrophy: signal change in the thalami and basal ganglia and thin, hyperintense, periventricular high-signal rims of white matter (Riikonen et al., 2000). A progressive diffuse brain atrophy on MRI is seen in children during the first 4 years of life which then usually stabilizes. MR spectroscopy shows a decrease in the N-acetyl aspartate (NAA) peak and increased choline, however, with the rapid progression of the disease, all peaks completely disappear by the age of 6 years (Vanhanen et al., 2004).
Neurophysiological findings in CLN1 disease are non-specific and include decreased reactivity of the posterior dominant rhythm to eye opening and eye closure, loss of sleep spindles by the age of 2 and an evolution towards an isoelectric electroencephalography (EEG) after the age of 3, which parallels the neuronal degeneration and brain atrophy.
CLN1: Differential diagnosis
Differential diagnosis should include other progressive neurodegenerative disorders with onset from birth to age 2 years: Rett syndrome, hexosaminidase A deficiency, leukodystrophies, peroxisomal disorders, Niemann-Pick disease types A and B, and Leigh syndrome. While some of these disorders are associated with cortical blindness, retinal involvement is rarely seen (Mole & Williams, 2013).
CLN1: Treatment
Treatment of CLN1 disease is symptomatic. As the enzyme cleaves fatty acid thioesters in plasma membranes, it was suggested that the drug cysteamine, a simple aminothiol used for the treatment of cystinosis, may have some effect. A clinical trial in children did not show any significant improvements (Levin et al., 2014) and in vitro studies also cast doubt on this concept (Lu & Hofmann, 2006). A phase I trial of intracerebral injection of human foetal neuronal stem cells has been performed (Guillaume et al., 2008; Mole, 2014) and enzyme replacement therapy has been considered (Lu et al., 2010).
CLN2
CLN2: Genetics
CLN2 encodes the lysosomal enzyme tripeptidyl peptidase (TPP1), a member of the serine carboxyl proteinase family (Rawlings & Barrett, 1999). This group of enzymes removes tripeptides from the N termini of small polypeptides such as the subunit c of mitochondrial ATP synthase. To date, more than 109 mutations have been described, the two most common being the splice site mutation c.509-1G>C and the nonsense mutation p.Arg208* resulting in broadly similar clinical phenotypes (Mole et al., 2005). The majority of the protein component of the storage bodies is subunit c of mitochondrial ATP synthase, as well as low amounts of saposins A and D (Lake & Hall, 1993; Jalanko & Braulke, 2009).
CLN2: Clinical presentation
Classic late-infantile CLN2 disease presents around the third year of life, with intractable epilepsy and an arrest of cognitive development. Myoclonus and ataxia are commonly seen early in the course, followed by progressive cognitive and motor decline. Retinopathy is often not prominent early in the course and may be overlooked after progression to more severe neurological deficits. Spasticity, truncal hypotonia, loss of head control, near-continuous myoclonus, frequent seizures, and an extended vegetative state are characteristic, until death in early adolescence. Death is often due to aspiration pneumonia. A few cases presenting late (8 years) have been reported, exhibiting slow regression with death as late as 40 years of age (Sleat et al., 1999).
CLN2: Diagnosis
Historically, diagnosis was made based on the presence of clinical features and the ultrastructural demonstration of curvilinear bodies. However, the diagnosis can now be reached rapidly and specifically by demonstrating a lack of TPP1 activity using blood, skin biopsy, saliva, or dried blot spot.
Brain MRI in CLN2 disease shows progressive cerebral atrophy that predominates in the infratentorial region. Hypointense thalami on T2-weighted images were also reported (Seitz et al., 1998). MR spectroscopy demonstrates a reduction in the NAA peak and an increase in myo-inositol and glutamate/glutamine in the white matter (Seitz et al., 1998).
EEG includes characteristic occipital spike responses to slow flash (1-2 Hz) stimulation which precede the onset of seizures and which increase as the disease progresses (figure 3). Electroretinogram is diminished even before noticeable visual loss (Wisniewski et al., 1998; Wisniewski et al., 2001; Goebel & Wisniewski, 2004). Visual evoked potentials are also enhanced at the onset of the disease.
CLN2: Differential diagnosis
Other progressive neurological diseases with onset from ages 2 to 4 years should be considered, including epileptic encephalopathies, lysosomal storage disorders, mitochondrial diseases, and leukodystrophies. Other rarer NCL variants, such as CLN5, CLN6, CLN7 and CLN8 diseases, should also be considered if TPP1 activity is normal.
CLN2: Treatment
Treatment is symptomatic. An experimental treatment approach uses intracerebral injection of viral vectors containing normal coding segments of the CLN2 gene. In a mouse model of CLN2 disease, this procedure resulted in cerebral enzyme expression, reduced brain pathology, and increased survival. A small number of human patients have recently been treated in the same way (Worgall et al., 2008), but the trial was too small to determine efficacy. A phase I trial of intracerebral injection of human foetal neuronal stem cells has been performed (Mink, 2010). Enzyme replacement therapy was efficacious in mice and dogs (Passini et al., 2006; Chang et al., 2008; Whiting et al., 2014) and a phase 1 trial is well underway in Europe (Biomarin).
CLN3
CLN3: Genetics
CLN3 encodes a membrane protein of unknown function. Whilst generally considered to be present predominantly in the endolysosome system, this protein has been reported to localize to membrane lipid rafts in synaptosomes, Golgi, and the cell membrane, as well as in mitochondria (Phillips et al., 2005). Currently more than 57 mutations have been characterized in the CLN3 gene. Most cases world-wide, which can be traced to a northern European origin, are due to a common ancestral 1-kb deletion founding mutation (Munroe et al., 1997) shown to retain partial activity (Kitzmuller et al., 2008).
Numerous roles have been attributed to the gene product of CLN3, and much work is needed to reconcile these disparate functions. In mitochondria, the gene product of CLN3 was suggested to aid the processing of mitochondrial membrane proteins, such as ATPase subunit c, which accumulates in this condition as a result of synaptic vesicle transport (Margraf et al., 1999). The CLN3 gene product has been implicated in the regulation of lysosomal pH, transport of basic amino acids into the lysosome, and lysosomal size (Golabek et al., 2000; Holopainen et al., 2001; Ramirez-Montealegre & Pearce, 2005). An antiapoptotic role has been ascribed to the gene product of CLN3; the C-terminus appears to participate in cell cycle regulation, and mutations of this region result in slow growth and increased apoptosis (Puranam et al., 1999). CLN3 knockout mice show neutralizing antibodies against glutamic acid decarboxylase (GAD65), suggesting that an autoimmune response against GAD65 might contribute to preferential loss of GABAergic neurons in this disease. However, it is not understood whether these autoantibodies contribute to the pathogenesis or whether they are secondary entities arising during neurodegeneration, although their presence may influence excitotoxic mechanisms (Chattopadhyay et al., 2002). Recently, an enzymatic function has been associated with the CLN3 gene product, namely palmitoyl-protein D-9 desaturase activity (Narayan et al., 2006). Another group showed a correlation between CLN3 expression and the synthesis of bis monoacylglycerol phosphate (BMP) and suggested that the CLN3 gene product may play a role in the biosynthesis of BMP (Hobert & Dawson, 2007).
CLN3: Clinical presentation
Classic juvenile CLN3 disease presents between the ages of 4 to 8 years (mean of 5 years) with a progressive loss of vision due to retinal degeneration, followed by progressive dementia. Ocular pathology is initially a pigmentary retinopathy often misdiagnosed as retinitis pigmentosa or cone dystrophy. During adolescence, epilepsy and extrapyramidal/parkinsonian signs (rigidity, hypokinesia, shuffling gait, impaired balance) are more prominent. Neuropsychiatric symptoms, such as anxiety and aggression, are common (Marshall et al., 2005). The clinical course is variable but inexorably progressive toward death in the second or third decade. CLN3 disease may present in adulthood with visual failure sometimes later accompanied with heart failure (Eksandh et al., 2000).
CLN3: Diagnosis
Ultrastructurally, CLN3 disease cases exhibit fingerprint profiles. These may be the only apparent feature within the lysosomal residual body, or may occur in conjunction with curvilinear or rectilinear profiles, or as a small component within large membrane-bound lysosomal vacuoles. The diagnostic hallmark of this frequent NCL type is conspicuous vacuoles in the cytoplasm of lymphocytes which are detectable on a regular blood smear (Anderson et al., 2005). Diagnosis is based on clinical suspicion, the presence of vacuolated lymphocytes, and ultrastructural studies or combined with genetic testing.
Brain MRI shows cerebral and cerebellar atrophy in the later stages (age > 15 years) and is normal before the age of 10 years. MR spectroscopy has not shown specific abnormalities.
The EEG shows non-specific progressive background disorganization and spike-and-slow-wave complexes. The predominant seizure type is generalized tonic-clonic, however, partial complex seizures can occur as well. Enhanced somatosensory evoked potentials may be seen, supporting the presence of a myoclonic component even before myoclonus is clinically present.
CLN3: Differential diagnosis
Differential diagnosis is limited given that juvenile CLN3 disease has a unique presentation. Peroxisomal, mitochondrial and other lysosomal disorders that are associated with retinopathy can be considered.
CLN3: Treatment
Treatment is symptomatic. Autoimmunity against GAD65 has been used as the basis for investigation of immunomodulatory treatments (Pearce et al., 2004).
CLN4
CLN4: Genetics
The CLN4 gene encodes DNAJC5, which underlies the autosomal dominant adult form of NCL, known as “Parry disease”. The gene symbol CLN4 was also used in the past to account for a heterogeneous group of adult forms of NCL which were recessively inherited (collectively recognised as Kufs disease) without known genetic loci at that time. Some of these forms were later identified as being secondary to mutations in CLN6 and CLN13.
Other NCL genes that may present with adult-onset are CLN1, CLN5, CLN10, CLN10 and CLN13 (table 2). While for most NCLs there is a specific phenotype associated with the loss of function of a particular CLN gene, for some NCLs that arise from mutations that have an incomplete effect of the gene function, the associated phenotypes are protracted or have a later age of onset (reviewed in Mole & Cotman [2015]).
CLN4: Clinical presentation
Adult-onset NCL can present with two different clinical phenotypes: Kufs type A with marked myoclonus, progressive epilepsy, dementia and ataxia; and Kufs type B, marked by behavioural changes and dementia, as well as peculiar facial dyskinesia. Vision is not impaired in primary adult forms of NCL.
CLN4: Diagnosis
Ultrastructural patterns include granular, curvilinear, or fingerprint profiles in different cell types and organs of the same patient, or a combination of patterns. Vacuolated lymphocytes were not reported.
CLN4: Differential diagnosis
All NCL with possible onset in adulthood should be included in the differential diagnosis. The pattern of inheritance may point towards an autosomal recessive or autosomal dominant form.
CLN4: Treatment
Treatment is symptomatic.
CLN5
CLN5: Genetics
CLN5 encodes a soluble protein that is directed to the lysosome. It is reported to interact with the gene products of CLN2 and CLN3 (Vesa et al., 1995). These observations suggest that there may be common molecular pathways or important interactions between pathways in various types of NCLs. Currently, more than 36 different mutations have been described in CLN5. The most common mutation, occurring in patients of Finnish origin, is a 2-base pair deletion in exon 4 (c.1175_1176delAT) that results in an early stop codon (p.Tyr392*).
CLN5: Clinical presentation
The first symptoms of the disease typically begin between 4 and 7 years of age (slightly older than the range in CLN2 disease), although cases are reported that present in adulthood. The usual course is motor clumsiness followed by progressive visual failure and blindness, dementia, motor decline, myoclonus, and seizures. The rate of progression is variable, but ultimately death occurs between the ages of 14 and 36 years (Mink, 2010). First reported in Finland, this type of NCL has recently been observed in many other European countries (UK, Czech Republic, Netherlands, Portugal, Italy), in North America (Canada, USA), South America (Argentina, Colombia), and other countries including Afghanistan and Pakistan. It should be considered in any exhaustive diagnostic approach of a patient with suspected NCL, especially with onset in late infancy but also up to adulthood (Santavuori et al., 1991).
CLN5: Diagnosis
Ultrastructurally, lipopigments are distributed in the central nervous system and extracerebrally, and include fingerprint bodies, curvilinear profiles, lamellar inclusions, and occasionally condensed fingerprint images associated with lipid droplets. The major stored material is subunit c of the mitochondrial ATP synthase (Tyynela et al., 1997). Similar to CLN3-defective fibroblasts, CLN5-deficient fibroblasts also exhibit elevated intralysosomal pH (Kyttala et al., 2006).
Brain imaging shows prominent cerebellar atrophy and in addition, on T2-weighted images, the thalamic signal intensity is low compared to that of the caudate, while increased signal intensity is seen in the periventricular white matter and the posterior limb of the internal capsule (Autti et al., 1992). Neurophysiological examination shows giant visual evoked potentials, exaggerated somatosensory potentials, and occipital spikes in response to photic stimulation, similar to CLN2. Ultimately, CLN5 can only be confirmed by DNA analysis.
CLN5: Differential diagnosis
The clinical presentation with dementia, motor clumsiness and visual failure is strongly suggestive of a neurodegenerative disease. The probability of NCL is further supported by the electrophysiological and imaging studies.
CLN5: Treatment
Treatment is symptomatic. The experimental finding that at least a portion of the CLN5 gene product is trafficked via the manose-6-phosphate pathway (Sleat et al., 2005) means that therapeutic approaches that depend upon cross-correction, including enzyme replacement therapy, gene therapy, and stem cell transplantation are likely to be tested in the near future (Selden et al., 2013).
CLN6
CLN6: Genetics
CLN6 encodes a protein of unknown function with seven transmembrane domains localizing to the endoplasmic reticulum (ER) (Sharp et al., 2003; Heine et al., 2004). Currently, more than 68 different mutations have been described in CLN6 disease. Patients are found all over the world, with particular concentrations in Costa Rica and Portugal, arising from a founder effect, as well as a range of mutations in Turkey and Newfoundland.
CLN6: Clinical presentation
Age at onset of CLN6 disease straddles the ages at onset of CLN1, CLN2, and CLN3 diseases, ranging from 18 months to 8 years, with the majority between 3 and 5 years. Early visual failure occurs in about 50 per cent of patients. The most prominent symptoms are motor impairment, including developmental delay, dysarthria, and ataxia. Seizures occur in the majority of patients, and usually begin before age 5 years. Deterioration is rapid after diagnosis and most children die between the ages of 5 and 12 years.
CLN6: Diagnosis
Diagnosis is based on clinical suspicion and genetic testing. Ultrastructurally, a mix of rectilinear profiles and fingerprint profiles are seen. The stored material contains subunit c of mitochondrial ATP synthase (Elleder et al., 1997). There is marked neuronal loss in layer V of the cerebral cortex and the extent of cerebral atrophy in CLN6 patients has been shown to be proportionate to the duration of symptoms based on post-mortem data (Elleder et al., 1997).
MR imaging shows progressive cerebral and cerebellar atrophy. As in CLN2 disease, EEG shows progressive background slowing and high-amplitude discharges in the posterior head regions in response to the photic stimulation.
CLN6: Differential diagnosis
Other NCL variants should be considered in the differential diagnosis of CLN6 disease. CLN1, CLN2 and CLN10 diseases can be excluded easily by enzyme analysis of PPT1, TPP1 and CTSD. Lymphocyte vacuolation, a hallmark of CLN3, is not seen in CLN6 disease.
CLN6: Treatment
Treatment is symptomatic. As the function of the CLN6 protein remains unknown, no experimental therapeutic studies have yet been initiated.
CLN7
CLN7: Genetics
The CLN7 gene product belongs to the large major facilitator superfamily (MFS) that transports specific classes of substrates, including sugars, drugs, inorganic and organic cations, and various metabolites (Jalanko & Braulke, 2009). CLN7 is also referred to as MFSD8. CLN7 is localized to lysosomes (Kousi et al., 2009). At present, more than 31 disease-causing mutations have been reported.
CLN7: Clinical presentation
Age at onset is usually between 2 and 7 years. Psychomotor regression or seizures are the initial presenting signs. Progressive cognitive and motor deterioration, myoclonus, personality changes and blindness occur later. The disease has a rapidly progressing course. A Rett syndrome-like onset has also been reported (Craiu et al., 2015).
CLN7: Diagnosis
Ultrastructural examination reveals both fingerprint patterns and rectilinear patterns. This form can be diagnosed by ultrastructural pathological analysis of peripheral lymphocytes where dense fingerprint profiles are observed. Vacuolations are not usually present in lymphocytes. Diagnosis is based on clinical suspicion and genetic testing.
Brain MR studies are abnormal from the early stages of the disease and show progressive cerebral and cerebellar atrophy, thinning of the corpus callosum, and hypointensity of the thalami on T2-weighted images.
Neurophysiological studies show diffuse background slowing of the EEG with occipital spikes, more prominent during sleep, which may evolve into electrical status epilepticus during slow-wave sleep.
CLN7: Differential diagnosis
Differential diagnosis includes mainly CLN3 and CLN6 diseases. Condensed fingerprint profiles in the lymphocytes and the absence of vacuolation is characteristic for CLN7 disease.
CLN7: Treatment
Clinical management is supportive. No experimental therapeutic trials have been initiated so far.
CLN8
CLN8: Genetics
CLN8 encodes a polytopic membrane protein that is localized to the ER and shuttles between the ER and ER-Golgi intermediate complex. The exact function is unknown, but it belongs to the TRAM-Lag1p-CLN8 (TLC) family of proteins, which are suggested to have roles in biosynthesis, metabolism, transport, and detection of lipids (Jalanko & Braulke, 2009). More than 24 mutations have been described.
CLN8: Clinical presentation
Depending on the mutation, CLN8 disease presents with childhood-onset (5-10 years), intractable epilepsy, followed by progressive cognitive decline or mild developmental delay in late infancy, followed by a florid PME with progressive myoclonus (Minassian et al., 2016), seizures, retinopathy, and psychomotor regression starting between 3 and 6 years. This typical late-infantile NCL phenotype leads to loss of vision (Topcu et al., 2004).
In the very specific subtype of Progressive Epilepsy with Mental Retardation or Northern Epilepsy, caused by a defined missense mutation p.Arg24Gly, age at onset is 5 to 10 years and seizures are the first symptom. All patients have generalized tonic-clonic seizures with frequent episodes of status epilepticus. As patients pass through puberty, the frequency of seizures decreases but progressive dementia and motor impairment continues (Ranta & Lehesjoki, 2000). Patients with Northern Epilepsy may survive until 50-60 years of age.
CLN8: Diagnosis
Diagnosis is based on clinical suspicion and genetic testing. GROD, curvilinear, and fingerprint profiles have been reported on electron microscopy, in various tissues, including lymphocytes, however, the stored material consists mostly of subunit c of mitochondrial ATP synthase.
MR studies show progressive cerebral and cerebellar atrophy with thinning of the corpus callosum. Neurophysiological testing is similar to other NCLs. Diagnosis can only be confirmed by DNA analysis.
CLN8: Treatment
Treatment is supportive, and no experimental therapeutic trials have been attempted so far.
CLN9
CLN9: Genetics
CLN9 has been proposed as a specific NCL entity, but no gene has yet been identified. Fibroblasts from affected families have a distinctive phenotype (rapid growth, sensitivity to apoptosis, manifestation of a cell adhesion defect, and reduced levels of ceramide, dihydroceramide, and sphingomyelin) (Schulz et al., 2004). Little is known of the function of the unidentified CLN9 protein.
CLN9: Clinical presentation
CLN9 is clinically indistinguishable from juvenile CLN3 disease, but perhaps with a more rapid course.
CLN9: Diagnosis
Ultrastructure is characterized by GRODs and curvilinear bodies. Diagnosis is one of exclusion. For patients presenting with typical features of CLN3 disease who have characteristic ultrastructural abnormalities, but no mutation in CLN3, possible CLN9 disease should be considered.
CLN10
CLN10: Genetics
The affected gene encodes cathepsin D (CTSD), a lysosomal enzyme thought to be important for neuronal stability (Siintola et al., 2006; Steinfeld et al., 2006), which is also secreted and exerts effects in the extracellular environment. More than 7 mutations have been described. Alterations in a macroautophagy-lysosomal degradation pathway appear to mediate neurodegeneration in this disease.
CLN10: Clinical presentation
CLN10 disease is characterized (in the congenital form) by primary microcephaly, neonatal (possibly already intrauterine) epilepsy, respiratory insufficiency, and rigidity. Death occurs within hours to weeks after birth. Late-onset forms of this NCL may be seen in juveniles and adults (Steinfeld et al., 2006). In one patient, missense mutations caused a childhood onset neurodegenerative disease with ataxia, retinopathy, severe cognitive decline, and apparently no seizures at age 17. The pathological correlate was GRODs (Steinfeld et al., 2006).
CLN10: Diagnosis
Diagnosis is based on clinical presentation and enzymatic testing for CTSD in fibroblasts or blood. Genetic testing for mutations in CLN10 is also available. On post-mortem examination, massive loss of neurons in the cerebral cortex, extensive gliosis, absence of myelin, and autofluorescent storage bodies with a GROD ultrastructure have been described.
CLN10: Treatment
Treatment is symptomatic and mainly targets quality of life.
CLN11
CLN11 disease is characterized by rapidly progressive visual loss due to retinal dystrophy, seizures, cerebellar ataxia, and cerebellar atrophy. Two disease-causing mutations, present as compound heterozygous in the GRN gene, have been reported in two Italian siblings from nearby villages in Lombardy, Italy. The transmission pattern of adult-onset NCL in the family was consistent with an autosomal recessive inheritance. Electron microscopic examination of a skin biopsy demonstrated numerous fingerprint profiles in membrane-bound structures in eccrine secretory cells and in endothelium, consistent with NCL. EEG results showed polyspike-wave discharges with a posterior emphasis, and MRI indicated cerebellar atrophy. Heterozygous mutations in this gene were previously known to cause frontotemporal lobe dementia (Smith et al., 2012).
CLN12
CLN12 disease was reported recently in a Belgian family. The index case had unsteady gait, myoclonus, and mood disturbance from age 11 to 13, progressing to clear extrapyramidal involvement with akinesia and rigidity, as well as dysarthric speech. There was no retinal involvement. Exome sequencing identified a single homozygous mutation in ATP13A2 that fully segregated with the disease within the family. Muscle biopsy showed numerous subsarcolemmal autofluorescence bodies with a fingerprint appearance under electron microscopy, suggestive of neurogenic muscular atrophy. Mutations in ATP13A2 are better known to cause Kufor-Rakeb syndrome (KRS), a rare parkinsonian phenotype with juvenile onset (Bras et al., 2012).
CLN13
Five disease-causing mutations have been reported in CLN3 (Smith et al., 2013). CLN13 disease is an adult-onset neuronal ceroid lipofuscinosis with cathepsin F (CTSF) deficiency, without vacuolated lymphocytes. The clinical phenotype (Kufs type B) is characterized by behaviour abnormalities and dementia, which may be associated with motor dysfunction, ataxia, extrapyramidal signs, and bulbar signs. Electron microscopy has showed fingerprint profiles in some cases. The protein product, CTSF, is part of the papain family of cysteine proteinases that represent a major component of the lysosomal proteolytic system.
CLN14
One disease-causing mutation has been reported in a Mexican family with vision loss, cognitive and motor regression, premature death, and prominent NCL-type storage material (Staropoli et al., 2012). The gene product, BTB/POZ domain-containing protein KCTD7, is a potassium channel tetramerization domain-containing protein 7. Other mutations in this protein cause infantile PME or opsoclonus-myoclonius ataxia-like syndromes.
Diagnostic approach of NCLs
NCL have a recognizable phenotype that correlates with the progressive grey matter neurodegenerative process involving the cortex, deep grey nuclei, cerebellum, and retina. The first approach to diagnosis should consider age at onset and type of clinical presentation, and has been well summarised recently (Schulz et al., 2013).
Presentation in a neonate with severe epilepsy and microcephaly should suggest CLN10 disease as a possible diagnosis. Enzyme testing for CTSD (CLN10) should be the first step. If this is negative, further or concurrent enzyme testing for PPT1 and TTP1 should be attempted before more invasive biopsies.
In young children (> 6 months) with otherwise unexplained epilepsy and developmental arrest, CLN1 and CLN2 diseases are the most likely considerations. If enzyme testing for PPT1 and TTP1 are negative and electron microscopy demonstrates typical storage material, genetic testing for CLN5, CLN6, CLN7/MFSD8, CLN8 and CLN14/KCTD7 should be considered.
A school-aged child presenting with rapid visual loss between ages 4 and 7 should be tested for CLN3 disease, first looking for lymphocyte vacuoles. If no lymphocyte vacuolization is present and testing for PPT1, TPP1 and CTSD is also negative, skin biopsy is indicated to assess if typical NCL storage material is present. If so, genetic testing for CLN5, CLN6, CLN7/MFSD8, CLN8, and CLN12/ATP13A2 is indicated.
The NCL variants CLN5, CLN6, CLN7, or CLN8 should be considered in a child who phenotypically and ultrastructurally has an NCL, but testing for the more common entities is negative.
In adults with non-specific mental, motor, or behavioural abnormalities in which NCL is suspected, the first line of investigation includes the enzymatic assays for PPT1, TTP1, CTSD and CTSF which, if normal, should prompt ultrastructural examination. If storage material is present, genetic testing for autosomal recessive (CLN6, CLN11/GRN, CLN13/CTSF) and autosomal dominant NCL (CLN4/DNAJC5) should be initiated, and if negative, all other remaining NCL genes should be investigated.
Generally speaking, enzyme testing should be performed first. Ultrastructural examination of skin or lymphocytes should be performed prior to gene sequencing, however, gene “chips” or other new DNA approaches that test concurrently for multiple NCL genes in the near future will reverse this approach. If there is no storage material, NCL is highly unlikely, although possible.
Symptomatic treatment for children with NCL
Patients with NCL require symptomatic treatment for a constellation of neurological manifestations, including seizures, sleep problems, extrapyramidal symptoms, behavioural problems, anxiety, and psychosis.
Routine medical management of children and young adults with complex neurological disability is relevant to all those affected by NCL, including clinical surveillance for sialorrhea, swallowing difficulties, gastroesophageal reflux, aspiration pneumonia, and X-ray surveillance of hips and spine.
Seizures may not require early treatment if generalized convulsions are rare and epileptic myoclonus is not obvious. As seizures progress, antiepileptic drugs of choice are valproic acid and lamotrigine, alone or in combination. However, lamotrigine may exacerbate seizures in CLN2 disease. Topiramate and levetiracetam are also effective. Benzodiazepines are useful in combination therapy, but they may cause problems due to sialorrhea. Carbamazepine, phenytoin, and gabapentin should be avoided as they may worsen myoclonic seizures. The ultimate goal must always be to improve the quality of life, and for this disease, the aim is not focussed on becoming seizure-free. Sleep disturbance is common and is likely to worsen with age. In general, a calm environment and set routines before going to bed are helpful. Benzodiazepines and sedatives are commonly used. Melatonin was found to have limited effect. Benzodiazepines may also benefit anxiety and spasticity. Trihexyphenydil improves dystonia and sialorrhea.
Emotional, behavioural and psychotic problems are common. Delusions and hallucinations may be managed using newer atypical neuroleptics, such as risperidone or olanzapine. If depression is thought to be the underlying problem, selective serotonin reuptake inhibitors may be considered which have been found to be beneficial. In all cases, medication should be kept to a minimum in order to avoid side effects or worsening disease progression.
Genetic counselling and testing of NCLs
The NCLs are inherited in an autosomal recessive manner with the exception of adult NCL, which can be inherited in either an autosomal recessive or an autosomal dominant manner. The parents of an affected child are obligate heterozygotes and are asymptomatic. The siblings of a proband have a 25 per cent chance of being affected, a 50 per cent chance of being an asymptomatic carrier, and a 25 per cent chance of being unaffected and not a carrier.
Prenatal testing is possible in high-risk pregnancies (if biochemical studies in the proband have revealed deficient activity of the enzymes CTSD, PPT1, or TPP1, or mutations defined in any NCL gene). In these instances, testing is performed on foetal cells obtained by chorionic villus sampling at 10-12 weeks of gestation or amniocentesis usually performed between 15-18 weeks of gestation (Mole & Williams, 2013).
Acknowledgements and disclosures
We would like to especially thank Dr. Cameron A. Ackerley for the micrographs used in the figures. We thank the many clinicians whose experience we have drawn upon and those scientists whose original work has not been cited due to lack of space. The authors are not aware of any conflicts of interest. D.A.N. is supported through the Canadian League against Epilepsy Fellowship Award and University of Toronto Postgraduate Research Awards (Chisholm Memorial Fellowship, Elizabeth Arbuthnot Dyson Fellowship and the Joseph M. West Family Memorial Fund). B.A.M. holds the Michael Bahen Chair in Epilepsy Research and is supported by the Ontario Brain Institute and Genome Canada.
None of the authors have any conflict of interest to disclose.
The present manuscript is part of a Epileptic Disorders/Mariani Foundation supplement on Progressive Myoclonus Epilepsies, downloadable as a whole by visiting www.epilepticdisorders.com.