Epileptic Disorders
MENUNeonatal hyperekplexia with homozygous p.R392H mutation in GLRA1 Volume 16, issue 3, September 2014

Hyperekplexia (startle disease) is a rare non-epileptic neurogenetic disorder with both autosomal-dominant and autosomal-recessive inheritance, affecting three genes (GLRA1, GLRB, and SLC6A5) of the inhibitory glycinergic system (Rees et al., 1994; Rees et al., 2006; Harvey et al., 2008; Chung et al., 2010; Chung et al., 2013). Sporadic cases are more prevalent than familial cases but all present with clinical symptoms including exaggerated startle responses and neonatal hypertonia, with apnoea events in 56% of cases (Thomas et al., 2013). The diagnosis is usually straightforward, once clinical features are recognised.
The aim of this case report is to draw attention to this unusual clinical entity which is often misdiagnosed in neonates as congenital tetanus or a convulsive disorder. An early diagnosis is crucial to prevent the risk of sudden death or learning disabilities from apnoea or choking on food.
Case study
We present the case of a male newborn, admitted on the second day of life to our neonatal intensive care unit, for management of suspected convulsive seizures.
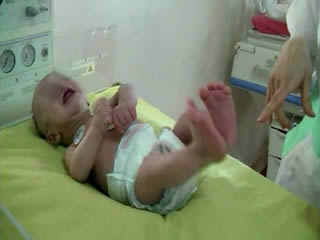
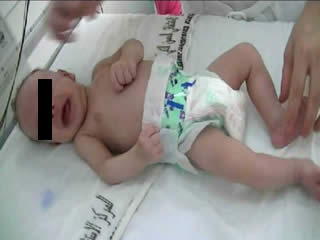
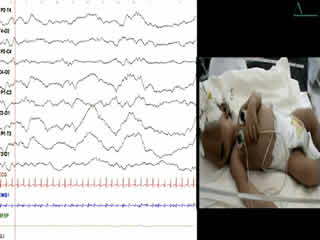
He was the first child of a consanguineous couple. Pregnancy and delivery showed no pathological events. The birth weight was 3,400 g. No complications were noted immediately after birth, with Apgar scores of 9 and 10 at 1 and 5 minutes, respectively. There was no family history of neurological disease. At 6 hours, he began to have frequent episodes of severe tonic movements accompanied with apnoea. On examination, there were no dysmorphic features. Head circumference was normal. The anterior fontanel was flat and of normal size. He was alert. Tone was diffusely and symmetrically increased. The baby presented acute, massive, and sustained episodes of stiffening of the trunk and limbs with severe bradypnoea (video sequences 1 and 2) and high-frequency trembling (video sequence 3) with clenching of the fists, during which time consciousness was unaltered, however, voluntary movements were impossible. Such episodes occurred in response to sudden noise and tactile stimuli. Initially, phenobarbital had been prescribed (20 mg/kg) due to a suspicion of neonatal seizures. However, the attack frequency remained the same. He was suspected to have infectious encephalitis or congenital tetanus which were ruled out by negative clinical history, examination, and direct laboratory testing. The newborn had normal serum levels of calcium, magnesium, sodium, potassium, glucose, urea, and creatinine. Cranial ultrasound and CT were normal. A cerebrospinal fluid study revealed no evidence of infection.
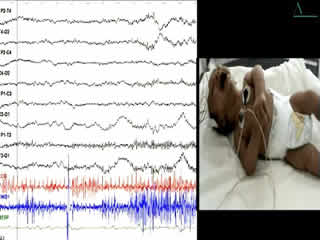
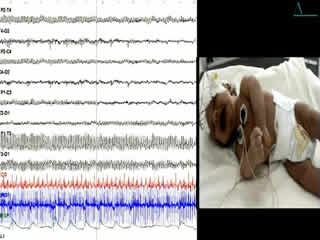
During the second week of hospitalisation, a startle reaction was observed after gently flicking the nose, with generalised flexor spasm and anxious staring without habituation (video sequence 4). Interestingly, hypertonia diminished during feeding and was absent during sleep. The apnoea and spasms could be stopped by forced flexion of the head and legs towards the trunk; the Vigevano manoeuvre (video sequence 5). A video-EEG was performed and the tracing showed acute muscle artefact and rapidly repeated giant compound muscle action potentials (CMAP) during attacks; the tracing was otherwise normal. Although there were no other similar cases in the family, a diagnosis of hyperekplexia was suggested. On the tenth day of life, clonazepam was begun, with immediate improvement with regards to tremulousness, hypertonicity, and especially the excessive response to facial stimulation. Feeding was normal. The infant was discharged from the hospital five days later and the mother was informed about how to stop an apnoea attack. He still had exaggerated startle reaction at the age of 6 months and he remained slightly hypertonic, though this had improved. His growth and development were normal.
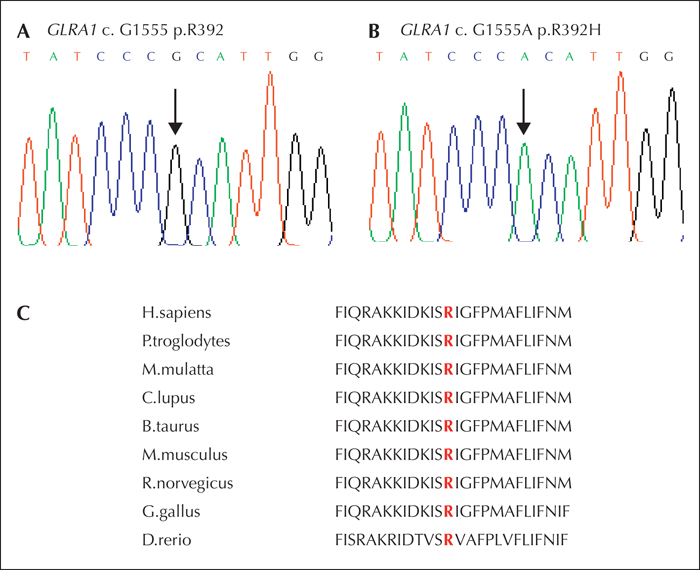
DNA from the index case was submitted to the Neurology and Molecular Neuroscience team at Swansea University, UK. Using multiplex PCR of the three hyperekplexia genes, a homozygous change in the GLRA1 gene was discovered (figure 1). This recurrent mutation was a homozygous G->A nucleotide change at position 1555 of the cDNA sequence (c.G1555A) generating an arginine to histidine substitution at amino acid position 392 (p.R392H). The missense mutation affects a highly conserved amino acid, located in the intracellular loop at the border with transmembrane domain 4 (TM4).
Discussion
Startle disease (or hyperekplexia) is a neurological disorder characterised by hypertonia, tremor, and exaggerated response to sudden tactile, auditory, and visual stimuli (Shahar and Raviv, 2004). The hallmark of this disorder is a failure of the startle response to habituate, and in many patients repeated stimuli elicit a greater response (sensitization). The disorder manifests immediately after birth by generalised hypertonia and stiffness attacks with preserved consciousness. Newborns are at risk of serious complications, such as brain damage and sudden death due to apnoea or aspiration; tactile stimuli of feeding can induce oropharyngeal incoordination (Sahar and Raviv, 2004). Such attacks may be resolved by a simple intervention, called the “Vigevano manoeuvre”; flexing of the head and limbs toward the trunk (Vigevano et al., 1989).
Clinical diagnosis of hyperekplexia is routine with prior awareness of the disease; it can be easily confirmed by a consistent generalised flexor spasm in response to tapping of the nasal bridge (Shahar et al., 1991). However, the startle and hypertonia episodes may be misdiagnosed as generalised tonic seizures (McMaster et al., 1999; Chen et al., 2007) or neonatal tetanus, and may be erroneously treated with antiepileptic drugs. Video-EEG or EEG with simultaneous observation by an experienced technician may be helpful in differential diagnosis.
Hyperekplexia may occur sporadically (as in our patient) or be inherited in an autosomal dominant or autosomal recessive manner. The primary cause of startle disease is defective inhibitory glycinergic neurotransmission (Chung et al., 2010; Chung et al., 2013). Mutations in five genes are linked to this disorder (Shiang et al., 1993; Rees et al., 1994; Rees et al., 2006; Harvey et al., 2008; Carta et al., 2012). Mutation in GLRA1, which encodes inhibitory glycine receptor (hGlyR) subunit α1, SLC6A5, which encodes the presynaptic sodium- and chloride-dependent glycine transporter 2 (GlyT2), and GLRB, which encodes glycine receptor subunit beta, are known to cause the hyperekplexia phenotype. Mutation in other genes represent very rare causes, however, patients often present with degenerative outcomes; such genes include GPHN (which encodes the glycinergic clustering molecule, gephyrin) and ARHGEF9 (which encodes collybistin). Patients with SLC6A5 and GLRB mutations were significantly more likely to have severe neonatal presentation and apnoea attacks (Rees et al., 2006; Thomas et al., 2013).
In this case study, a recurrent homozygous GLRA1 p.R392H mutation was detected which alters hGlyR expression and dynamics, and is unequivocally the major gene causing the phenotype and the basis for prenatal testing for further pregnancies. R392H has been identified in previous studies as part of a compound heterozygote (Vergouwe et al., 1999) and as a homozygous recessive mutation (Chung et al., 2010). Investigation of the functional effects of this mutation has revealed that GlyR trafficking is impaired in the presence of p.R392H, with reduced cell-surface expression of mutant GlyR α1 (Rea et al., 2002; Villmann et al., 2009). However, consanguinity may add further complications due to other allelic regions being reduced to homozygosity, and in the future, exome analysis may be required to detect the full genetic risk in this case.
Clonazepam is the first choice for treatment. Although the stiffness resolves by approximately three years of age, the exaggerated startle persists, resulting in frequent falls, injury, and social anxiety. Motor milestones are often mildly delayed, however, intellectual development is usually normal, although it can be complicated by consanguineous presentation.
In summary, hyperekplexia may present in the newborn period with hypertonia, hyperreflexia, and a characteristic exaggerated response to gently flicking the nose. This disorder is important to recognise, because of repeated neonatal and infantile apnoea and the increased risk of sudden infant death. Most infants respond to clonazepam for the treatment of hypertonia, and genetic analysis can help stratify outcomes, however, close follow-up is certainly recommended.
Acknowledgements and disclosures
The authors have no conflicts of interests to disclose.