Epileptic Disorders
MENUMyoclonus epilepsy and ataxia due to potassium channel mutation (MEAK) is caused by heterozygous KCNC1 mutations Volume 18, supplement 2, September 2016

Figures
- Key words: MEAK, KCNC1, progressive myoclonus epilepsies, myoclonus, seizures, potassium channel mutations, ataxia
- DOI : 10.1684/epd.2016.0859
- Page(s) : 135-8
- Published in: 2016
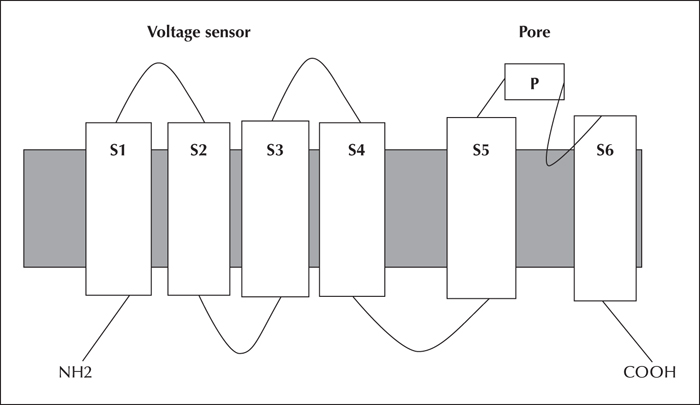
Progressive myoclonus epilepsy (PME) is a distinct group of seizure disorders characterized by gradual neurological decline with ataxia, myoclonus and recurring seizures. There are several forms of PME, among which the most recently described is MEAK - myoclonus epilepsy and ataxia due to potassium channel mutation. This particular subtype is caused by a recurrent de novo heterozygous mutation (c.959G>A, p.Arg320His) in the KCNC1 gene, which maps to chromosome 11 and encodes for the Kv3.1 protein (a subunit of the Kv3 subfamily of voltage-gated potassium channels). Loss of Kv3 function disrupts the firing properties of fast-spiking neurons, affects neurotransmitter release and induces cell death. Specifically regarding Kv3.1 malfunctioning, the most affected neurons include inhibitory GABAergic interneurons and cerebellar neurons. Impairment of the former cells is believed to contribute to myoclonus and seizures, whereas dysfunction of the latter to ataxia and tremor. Phenotypically, MEAK patients generally have a normal early development. At the age of 6 to 14 years, they present with myoclonus, which tends to progressively worsen with time. Tonic-clonic seizures may or may not be present, and some patients develop mild cognitive impairment following seizure onset. Typical electroencephalographic features comprise generalized epileptiform discharges and, in some cases, photosensitivity. Brain imaging is either normal or shows cerebellar atrophy. The identification of MEAK has both expanded the phenotypic and genotypic spectra of PME and established an emerging role for de novo mutations in PME.