Epileptic Disorders
MENULafora disease Volume 18, supplement 2, September 2016

Figures
-
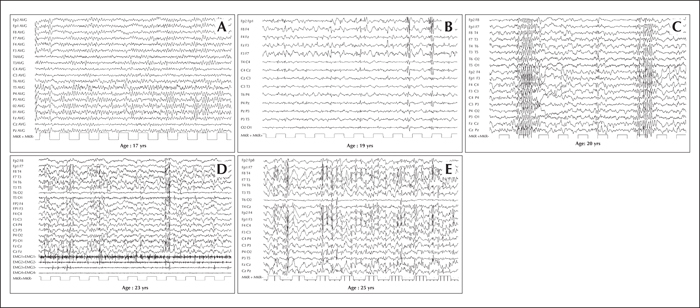
Figure 1 -
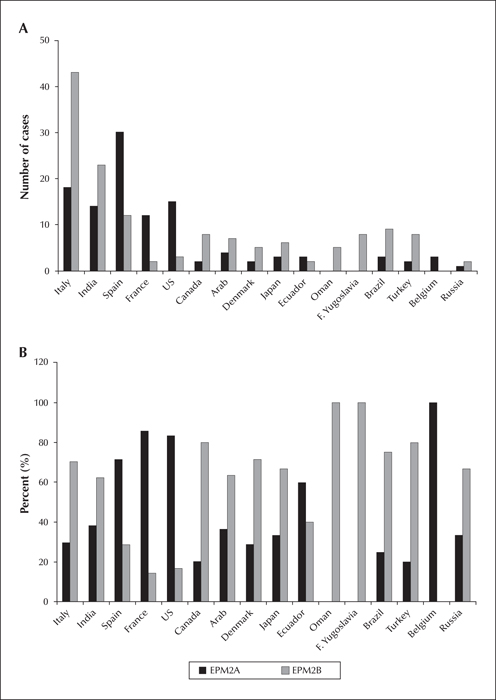
Figure 2 -
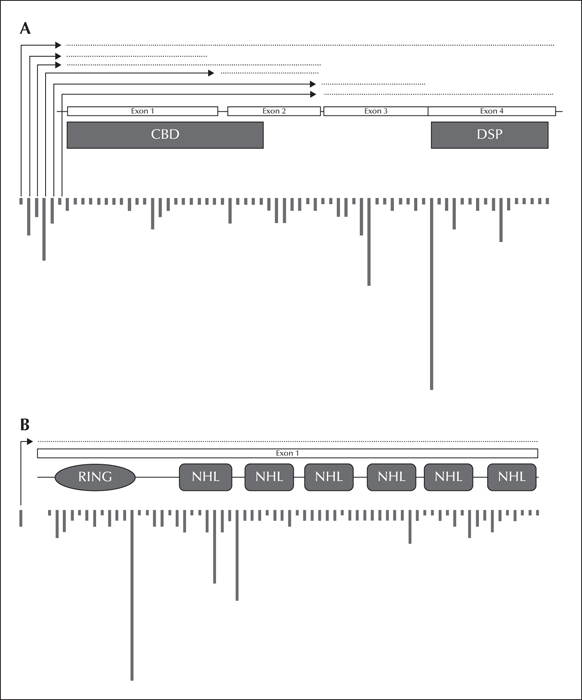
Figure 3 -
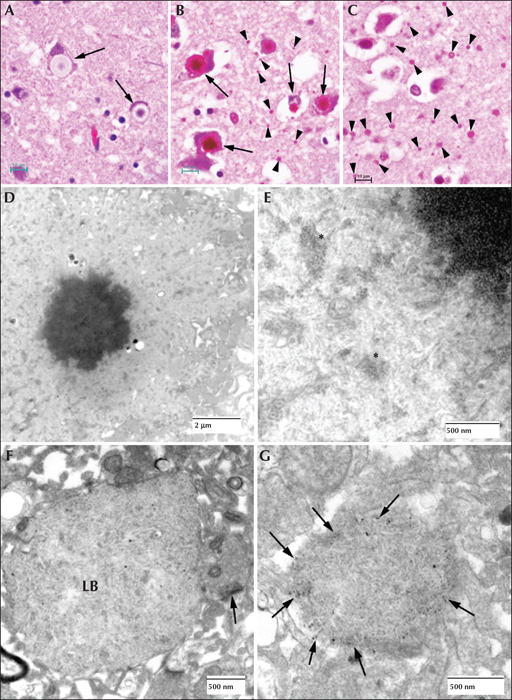
Figure 4 -
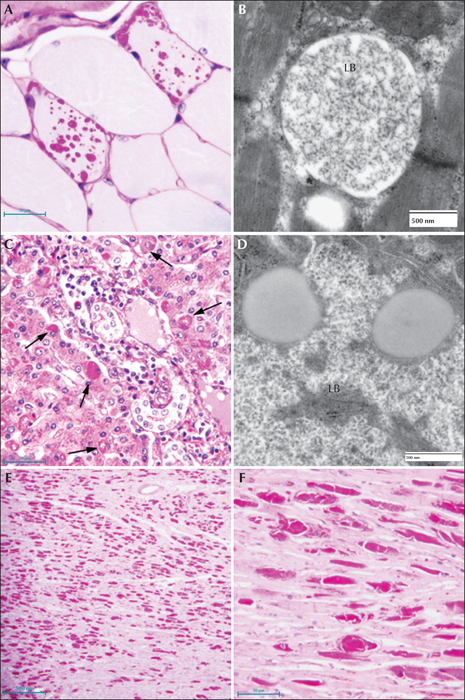
Figure 5 -
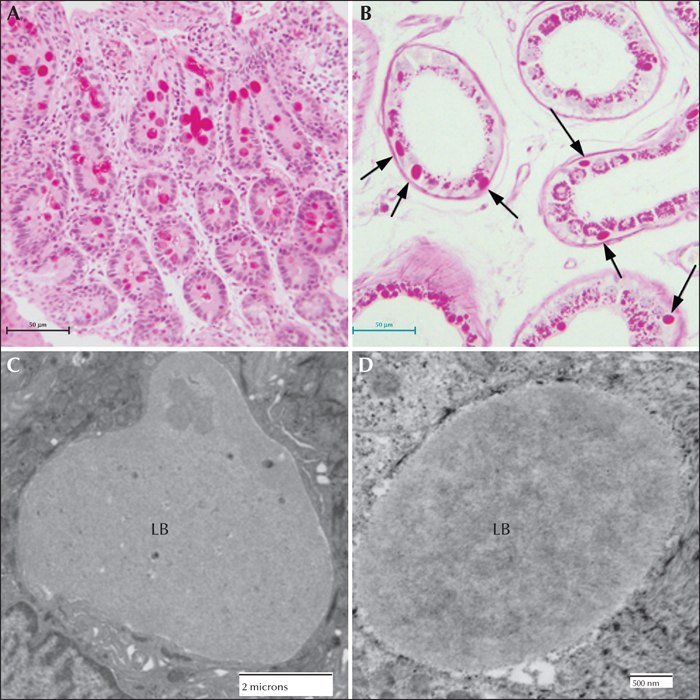
Figure 6 -
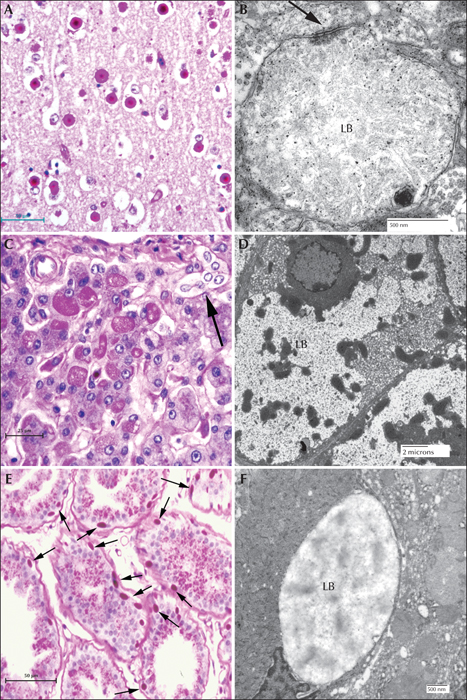
Figure 7 -
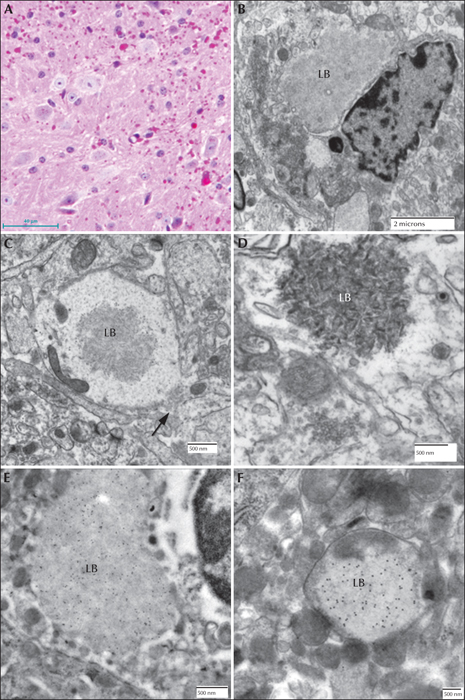
Figure 8 -
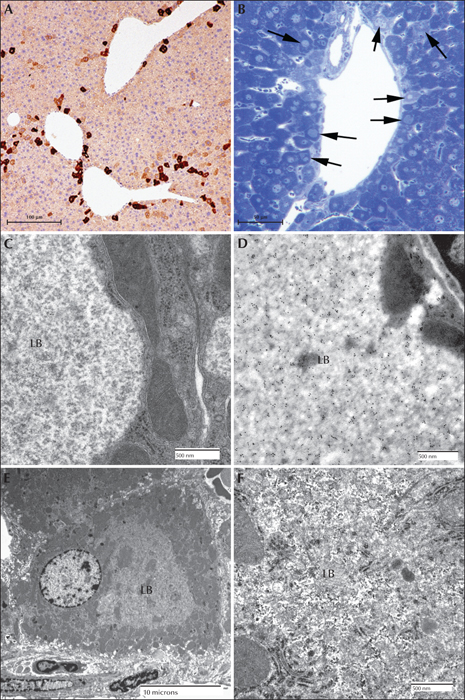
Figure 9 -
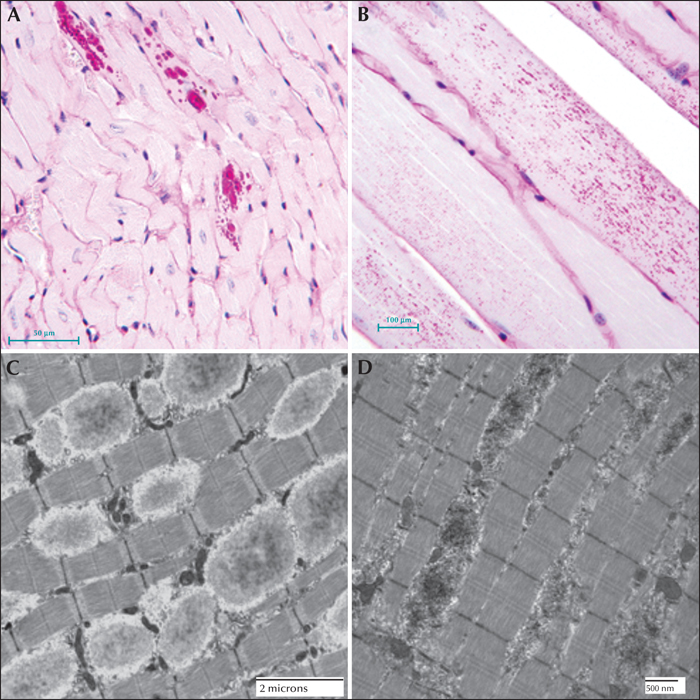
Figure 10 -
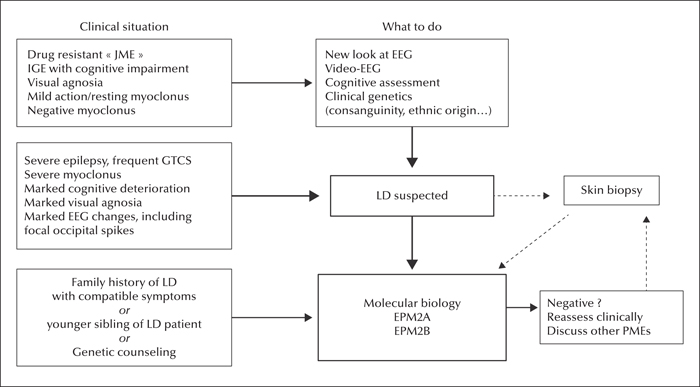
Figure 11 -
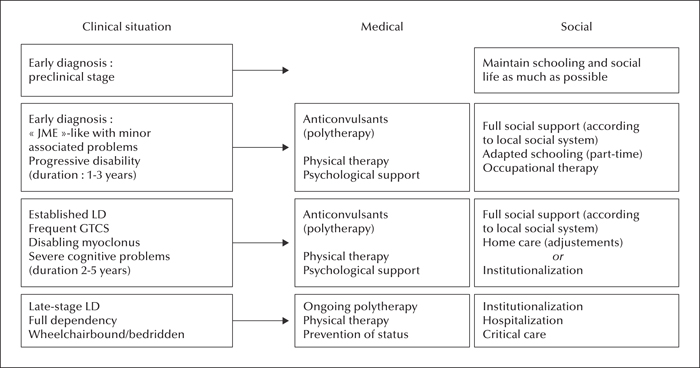
Figure 12
Tables
Lafora disease (LD, OMIM# 254780) is an autosomal recessive progressive myoclonus epilepsy (PME) (Minassian et al., 2016), first described in 1911. It is particularly frequent in Mediterranean countries (Spain, Italy, France), Northern Africa, the Middle East, and in some regions of Southern India where a high rate of consanguinity is present (Minassian, 2001; Striano et al., 2008). It can, nevertheless, be found in any population (Traoré et al., 2009), and particularly, as expected, with consanguinity.
LD classically starts in adolescence in otherwise neurologically normal individuals, usually with action and stimulus-sensitive myoclonus, as well as tonic-clonic, absence, atonic, and visual seizures. Neuropsychiatric symptoms, such as behavioural changes, depression and apathy, are also often present. Initial symptoms are followed by rapidly progressing dementia, refractory status epilepticus, psychosis, cerebellar ataxia, dysarthria, mutism, and respiratory failure which lead to death within about a decade (Minassian, 2001; Striano et al., 2008). LD is caused by mutations in the EPM2A orEPM2B (NHLRC1) genes, encoding the laforin dual specificityphosphatase and the malin ubiquitin E3 ligase, respectively, both involved in a complex and yet very incompletely understood pathway regulating glycogen metabolism (Minassian et al., 1998; Chan et al., 2003a, 2003b). To date, an additional gene, PRDM8, the mutation of which causes a variant of early childhood-onset phenotype in a single family, has been reported (Turnbull et al., 2012).
A distinctive pathology characterizes LD. Cells of various types exhibit dense accumulations of malformed and insoluble glycogen molecules, known as polyglucosans, which differ from normal glycogen due to the fact that they lack the symmetric branching that allows glycogen to be soluble. These polyglucosan accumulations are called Lafora bodies (LBs) and are profuse in all brain regions and in the majority of neurons, specifically in their cell bodies and dendrites (Minassian, 2001; Striano et al., 2008). Neuronal LBs localize in perikarya and dendrites but not in axons, possibly explaining the cortical hyperexcitability seen in LD. The neuropathology of LD patients and LD animal models is described in greater detail below.
Clinical features and diagnosis
The first symptoms of LD appear during late childhood or adolescence (range: 8-19 years; peak: 14-16 years). Characteristically, focal visual seizures are early manifestations and present as transient blindness, or simple or complex visual hallucinations. However, generalized seizure types, e.g. tonic-clonic, absence, or drop attacks, as well as myoclonus, occur soon after. The latter typically occurs at rest and increases with emotion, action, or photic stimulation. In many cases, the disease shows an insidious near-simultaneous, or closely consecutive, appearance of headaches, difficulties at school, myoclonic jerks, generalized seizures, and visual hallucinations (Minassian, 2001; Franceschetti et al., 2006; Striano et al., 2008). It is notable that not all visual hallucinations are epileptic in Lafora patients, as some respond initially to antipsychotic, rather than antiepileptic, medications (Andrade et al., 2005).
EEG abnormalities often precede clinical symptoms and initially consist of almost normal or slowed background (figure 1A) and generalized or focal paroxysmal activity (figure 1B), typically not accentuated by sleep. In particular, the occipital discharges on EEG, arising from a slowed posterior dominant rhythm are, in the proper clinical context, highly suggestive of the disease. Within a few years, a slowing of background activity becomes evident with frequent, superimposed bursts of diffuse epileptic discharges (figure 1C). In addition, positive or negative myoclonus (figure 1D) and marked photosensitivity (figure 1E) are prominent features.
Brain MRI is usually unremarkable at onset. In two reported cases (Jennesson et al., 2010), [18]fluorodeoxyglucose positron emission tomography (FDG-PET) revealed posterior hypometabolism early during the evolution of the disease. Electrophysiological investigations (to examine jerk-locked averaging, somatosensory evoked potentials, C-reflex, and visual evoked potentials) can reveal aberrant integration of somatosensory stimuli and cortical hyperexcitability (i.e. giant evoked potentials). Visual evoked potentials may show increased latencies or absence of response. Other findings obtained by transcranial magnetic stimulation indicate a complex circuitry dysfunction, possibly involving both excitatory and inhibitory systems (Canafoglia et al., 2010). A skin biopsy shows the presence of the characteristic periodic acid-Schiff (PAS)-positive glycogen-like intracellular inclusion bodies in the myoepithelial cells of the secretory acini of the apocrine sweat glands and in the eccrine and apocrine sweat duct cells (Andrade et al., 2003; Lohi et al., 2007). Electron microscopy confirms the presence of fibrillary accumulations, typical of polyglucosans. This diagnostic approach offers limited invasiveness and high sensitivity. Interpretation of the biopsy, however, requires expertise in distinguishing LB from normal polysaccharide contents of apocrine sweat glands, without which false-positive diagnosis is common. Genetic testing is crucial to confirm the diagnosis as it reveals mutations in the EPM2A or EPM2B gene in more than 95 per cent of patients (Minassian, 2001; Ganesh et al., 2006; Striano et al., 2008).
In the years following onset, symptoms of LD progress towards intractable action-sensitive and stimulus-sensitive myoclonus, refractory seizures, psychosis, ataxia, and dysarthria. As the disease progresses, the myoclonus remains asymmetric and segmental but becomes almost constant, and massive myoclonic jerks appear. At this stage, brain MRI may reveal mild cerebellar or cortical atrophy. Moreover, brain 1H MR spectroscopy shows metabolic changes of the cerebellum, basal ganglia, and frontal cerebral cortex (Villanueva et al., 2006; Pichiecchio et al., 2008). Subsequently, a rapidly progressive dementia with apraxia and visual loss soon appears. Speech becomes extremely difficult, and ataxia makes walking impossible. For many years, the patient struggles to maintain normal contact, with communication interrupted by extremely frequent myoclonic absence seizures (Minassian, 2001; Striano et al., 2008). Patients finally become totally disabled and bed-bound. Death usually occurs within 10 years from the onset, often during status epilepticus with aspiration pneumonia.
Differential diagnosis
At onset, LD may present with a clinical picture resembling an idiopathic generalized epilepsy, such as juvenile myoclonic epilepsy (Janz syndrome). In the early stages, drug resistance and a slow background with early disrupted sleep patterns should lead to a suspicion of LD. In some patients, the diagnosis is reached only after substantial follow-up of the patient. However, the main differential diagnosis concerns four other forms of PME: Unverricht-Lundborg disease (EPM1), the neuronal ceroid lipofuscinoses, myoclonic epilepsy with ragged red fibers (MERRF), and sialidosis (Berkovic et al., 1986; Minassian, 2001; Striano et al., 2008) (table 1). PMEs are a group of inherited neurodegenerative disorders characterized by progressively worsening myoclonus and epilepsy, variable neurological dysfunction (ataxia, dementia), and possible associated signs and symptoms. LD is one of the main teenage-onset PMEs. Age at onset, presenting symptoms, occurrence of occipital seizures, and the progressive and rapid course, together with the EEG features, are suggestive of LD, but the diagnosis is definitively confirmed by skin biopsy or genetic analysis. Unverricht-Lundborg disease (EPM1), caused by mutations of the cystatin B gene, is the closest differential. The age at onset is similar to that of LD. However, characteristically, action myoclonus and tonic-clonic seizures are more easily controlled. There is no specific pathology. Finally, cognitive impairment is not a distinctive symptom of Unverricht-Lundborg disease and is usually mild (Minassian, 2001; Striano et al., 2008). Neuronal ceroid lipofuscinoses are heterogeneous conditions characterized by ceroid-lipofuscin lysosomal storage occurring in different organs, including the central nervous system and skin (Nascimento et al., 2016). All NCLs exhibit ceroid lipofuscin accumulation, apart from LB, in various organs, including the skin. Most NCLs present in early childhood. A juvenile form of NCL (Batten's disease, involving the CLN3 gene) may have onset late enough to overlap with that of LD, but this form has a prolonged initial period of visual loss, resulting from retinal degeneration and mild seizures, which LD patients typically do not have. Some cases of the infantile form (Santavuori-Haltia disease, involving the CLN1 gene) present late as a result of particular mutations, and there is a very rare form of adult-onset NCL (Kufs disease; gene unknown), however, these diseases are excluded by the presence of LB. Mitochondriopathies, such as MERRF and MELAS, usually exhibit maternal mtDNA transmission. Clinical presentation may be heterogeneous and includes a wide range of associated symptoms, such as deafness, low stature, myopathy, lactic acidosis and optic atrophy, with variable prognosis. Sialidosis is an extremely rare lysosomal disease with cherry red spot maculopathy and elevated urine oligosaccharides (Berkovic et al., 1986).
Prognosis and evolution
The prognosis of LD is invariably progressive and fatal, leading to death 5-10 years after clinical onset. Genotype-phenotype correlations do not reveal substantial differences between patients carrying EPM2A and EPM2B mutations, but a few specific EPM2B mutations appear to correlate with a late onset and slow progressing LD (Minassian, 2001; Boccella et al., 2003; Franceschetti et al., 2006; Striano et al., 2008). At present, LD treatment remains palliative, with the best current therapies having limited success in the modulation of symptoms. Commonly used antiepileptic therapy for the management of myoclonus may improve symptoms during the early stages of the disease (Minassian, 2001; Striano et al., 2008). Hopefully, the work of many investigators, who are putting a massive effort into elucidating the pathophysiology, will result in focused treatments to control the deterioration of this otherwise devastating condition.
Epidemiology of Lafora disease and its genetic correlations
LD is a rare orphan disease. Based on all published reports of LD mutations, we estimate an overall frequency of ∼four cases per million individuals in the world. Over 250 patients and/or families have been described with LD (figure 2A). Of these, 42 per cent are caused by mutations in EPM2A and 58 per cent by EPM2B mutations (figure 2B). The ratio of EPM2A to EPM2B cases varies with population, with some regions having many more EPM2A cases than EPM2B, and vice versa, and this is, remarkably, not solely due to founder mutations (Gomez-Garre et al., 2000; Franceschetti et al., 2006). The most common EPM2A mutation is the R241X mutation, which accounts for approximately 17 percent of EPM2A-mediated LD. Large deletions make up 10-15 percent of EPM2A mutations, with the remainder ranging from those causing approximately eight percent of EPM2A-mediated cases of LD (i.e. R171H) to orphan mutations spread across the gene (figure 3A). For EPM2B, the two most common mutations are the missense mutation P69A and the frameshift mutation G158fs16 which affect ∼15 and ∼eight per cent of EPM2B-mediated LD, respectively. As with EPM2A, the remaining EPM2B mutations span the gene (figure 3B) and are rare, though some mutations (i.e. C26S and D146N) are more frequently observed. Because deletions can be overlooked using conventional sequencing techniques, it is critical to consider deletion/duplication analysis in any suspected LD patients in whom initial sequencing of EPM2A and EPM2B reveals no change.
Founder effects and recurrent mutations
Certain mutations appear to be specific to particular ethnic groups and/or geographical locations. For example, LD affects French Canadians from a geographically isolated area of eastern Quebec with an unusually high frequency, which is likely to be due to the ancestral EPM2B C26S mutation (Chan et al., 2003a, 2003b). One study has been published on LD in Oman, in which all cases in five separate, unrelated families resulted from a single ancestral mutational event in EPM2B (Turnbull et al., 2008). Interestingly, the EPM2A R241X mutation commonly found in individuals of Spanish descent resulted from both recurrent events and founder effects (Gomez-Garre et al., 2000; Ganesh et al., 2002a). At least five unique haplotypes are associated with this mutation, indicating that a minimum of five separate mutational events have led to the prevalence of the R241X mutation. Accordingly, EPM2A-mediated LD is more common than EPM2B-mediated LD in Spain. The second most common mutation in EPM2B, the missense P69A mutation, appears to have occurred from multiple mutational events similar to the EPM2A R241X mutation (Gomez-Abad et al., 2005). Only one common haplotype was found among eight patients analyzed with P69A mutations, strongly indicative of a recurrent event.
Phenotypic hetero- and homogeneity
Clinically, LD is a fairly homogenous disease with onset in adolescence and neurological decline soon after, but the timing and severity of symptoms can be variable, even within families. Both phenotypic heterogeneity and homogeneity have been noted in LD (see below) and with the large number of mutations in both EPM2A and EPM2B, genotype-phenotype correlations have been difficult to ascertain. Nonetheless, some associations between the two have been made. Gomez-Abad et al. identified three Arabic families with a complete deletion of the EPM2A gene (Gomez-Abad et al., 2007). Haplotype analysis and breakpoint mapping established a shared haplotype among the patients, suggesting a common ancestral origin for this mutation. Of the seven confirmed LD patients in the pedigrees, there was great variability in both disease onset and severity of symptoms, which was unexpected, due to the founder effect of the mutation. Clearly, even with the same ancestral mutation, phenotypic heterogeneity is common. This has been shown with other ancestral founder mutations, including the recurrent R241X EPM2A mutation (Gomez-Garre et al., 2000; Ganesh et al., 2002a). However, in a genetic isolate, the same ancestral founder mutation within a population resulted in phenotypic homogeneity (Turnbull et al., 2008). The uniformity of environmental and genetic background reinforces the idea that other genetic and/or environmental modifiers may influence the phenotypic spectrum in LD. Indeed, this has been shown within one family; an EPM2B patient harbouring a coding variant for the PPP1R3C gene, which encodes protein targeting to glycogen (PTG), exhibited a milder course of LD (Guerrero et al., 2011). The resulting conclusion from reports of both clinical homogeneity and heterogeneity in LD is that even among families, the disease course may or may not be identical and each LD patient is just as likely to have a clinically diverse, rather than classic, disease course.
Genotype-phenotype correlations
‘Atypical’ LD with early-onset childhood learning disabilities has been reported (Ganesh et al., 2002a; Annesi et al., 2004). Here, the authors showed a significant association with exon 1 mutations in EPM2A, linked to the development of learning disabilities prior to the onset of classic neurological symptoms of LD. Exon 4 mutations were mainly associated with classic LD with no childhood-onset educational difficulties. The authors suggest that exon 1 mutations can result in the complete loss of laforin function, whereas exon 4 mutations may preserve some of its functionality. However, other studies have not shown the same association, even with patients with the same or similar mutations in exon 1. Atypical LD with childhood learning difficulties, as observed by Ganesh et al. and Annesi et al., may prove to be a sub-syndrome of LD or could be due to as-yet-unknown genetic and/or environmental modifying factors (Lesca et al., 2010).
One study identified a family with two siblings with LD, one of whom presented with severe liver failure as an initial symptom (Gomez-Garre et al., 2007). Liver function in the sibling was abnormal, although the patient remained asymptomatic. Upon further follow-up, both developed classic LD, and mutation screening revealed a homozygous EPM2A R241X mutation in both siblings. Interestingly, the liver disease resembled type IV glycogen storage disease (GSD; due to GBE1 deficiency). The two diseases (type IV GSD and LD) share very similar features, namely the presence of polyglucosan bodies. This suggests a role for modifier genes in LD and the authors propose that these defects may lie in the same metabolic pathway, i.e. the glycogen metabolic pathway. The EPM2A R241X mutation is a prevalent mutation, and no other cases of hepatic failure have been identified in patients with this mutation. It is likely that other modifying factors led to this presentation, but the similarity to type IV GSD is intriguing and, as the authors of the study mention, certain modifiers may influence the severity of non-neurological symptoms in LD.
Some studies have indicated that patients with malin-mediated LD have a slightly milder disease course than patients with laforin-mediated LD (Baykan et al., 2005; Gomez-Abad et al., 2005; Singh et al., 2006). However, others found no change in severity between the two (Franceschetti et al., 2006; Lohi et al., 2007; Traore, 2009; Lesca et al., 2010; Brackmann et al., 2011). Conclusive evidence involving large-scale comparisons is lacking, but it is clear that there exists one relatively common, milder, EPM2B mutation, which may be skewing the comparative results in some analyses. Patients with either heterozygous or homozygous D146N mutation in EPM2B, in all cases in our experience and in the literature, have an atypical milder LD consisting of a later onset of symptoms, longer disease course, and extended preservation of daily living activities (Chan et al., 2003b; Baykan et al., 2005; Gomez-Abad et al., 2005; Francheschetti, 2006; Couarch et al., 2011). It is likely that at least some of the functionality of malin is preserved in patients with D146N mutations, as it has been demonstrated that the mutation preserves both the E3 ubiquitin ligase activity and a weak interaction with laforin (Solaz-Fuster et al., 2008). Clinical heterogeneity is common in LD, even among patients with allelic homogeneity and while it is true that certain mutations, such as D146N, cause a milder disease course, most cases of malin-mediated LD have a devastating disease course which is comparable to that of laforin-mediated LD.
The pathology of Lafora disease and Lafora disease animal models
The primary morphological change in LD is the deposition of polyglucosans, which consists of discrete deposits of fibrillary polysaccharides composed of poorly-branched glucose polymers (LBs). They are typically found in the brain, in the periportal hepatocytes of the liver, skeletal and cardiac myocytes, and in the eccrine duct and apocrine myoepithelial cells of the sweat glands (Sakai et al., 1970; Carpenter & Karpati, 1981a; Shirozu et al., 1985; Barbieri et al., 1987).
In the brain, the largest LBs tend to have two layers of various proportions in haemotoxylin and eosin (H & E)-stained preparations, the outer layer being pale and the core basophilic (figure 4A). These large bodies are usually found in neuronal perikarya. The ratio of perikaryal LBs to neurons is highest in the substantia nigra, followed by the dentate nucleus and some thalamic nuclei (figure 4B). They are rare or absent in the anterior part of the spinal cord. In the cerebral cortex, perikaryal LBs are rather scant, but a periodic acid Schiff (PAS) stain reveals numerous small LBs in the cortical neuropil (figure 4C), the majority being found in dendrites. These are commonly referred to as ‘dust-like’ in appearance. The PAS-positive material found in both perikaryal and ‘dust-like’ LBs, as well as LBs found in other tissues, are diastase-resistant, hence for all diagnoses of suspect LD patients, diastase-treated PAS (PASD) staining plays an integral role in the identification of the disease in both surgical biopsy and autopsy materials. Some patchy neuronal loss may be present in the later stages of the disease, but this is not a prominent feature (Sakai et al., 1970; Carpenter & Karpati, 1981a).
Electron microscopy confirms the presence of fibrillary accumulations typical of polyglucosans within the LBs. Using electron tomography, we were recently able to identify an ordered structure where individual polyglucosan fibrils bifurcate at a regular periodicity, ranging from 50-125 nm, depending on the tissue of origin (unpublished observation). This is especially true of the perikaryal and ‘dust-like’ LBs found in the brain, in skeletal muscle myocytes, and all of the LBs in affected sweat glands. Along with the filaments, there is often poorly-defined granular material within the LB itself. In two extremely well preserved brain biopsy specimens, we observed “glycogen-like” particles in the dendritic cytoplasm and the perykaryon of neurons in both perikaryal and ‘dust-like’ LBs (unpublished data) (figure 4D-G). Glycogen is rarely found in neurons because the sugars are rapidly metabolized. In skeletal muscle and cardiac myocytes, the fibrillary polysaccharide accumulates in membrane-bound spaces which are not lysosomal (Yokoi et al., 1975; Carpenter & Karpati, 1981a; Barbieri et al., 1987). There is variation in the morphology of the enclosed material, including some granular material, but the majority is fibrillar. Skeletal muscle can be used as a source for morphological diagnosis, but it is far from ideal, since the degree of involvement varies, and not all muscle fibre types are involved (Neville et al., 1974; Turnbull et al., 2011a) (figure 5A, B). This has been clearly demonstrated in a laforin knockout mouse model, and retrospective studies on human muscle biopsies are currently underway. Similar to the mouse, preliminary data suggest that humans do in fact produce LBs in type II fibres (Turnbull et al., 2011a).
Hepatic insufficiency is an early event in rare LD patients (Nishimura et al., 1980; Tomimatsu et al., 1985; Gomez-Garre et al., 2007). In the liver, PASD staining shows deposits of large portions of PASD-positive material in many of the periportal hepatocytes, confining the nucleus and other organelles to one side of the cell. Liver biopsy has been used for diagnosis, but the pathology is not specific to LD alone (Nishimura et al., 1980). Electron microscopy of periportal hepatocytes with LBs reveals lakes of loosely packed fibrillar material with some glycogen particles and/or rosettes within the LB. Although a few organelles such as mitochondria and peroxisomes can be found in the LB, the majority of the smaller organelles are confined to a thin layer of cytoplasm which is continuous with the thicker side of the cell which contains the nucleus, the majority of the endoplasmic reticulum, and the Golgi (figure 5C, D).
The occurrence of LBs in the sweat glands has become the gold standard in the pathological diagnosis of LD (Carpenter & Karpati, 1981b; Andrade et al., 2003). Skin is the most accessible of the affected tissues and biopsy procedures are the least invasive. LBs can be found in the eccrine duct cells, close to the secretory coil or close to the surface. They are extremely PASD-positive and are roughly the size of the cell nucleus. Under the electron microscope, LBs are almost ‘starch-like’ in appearance, however, this is not a diagnostic feature. The eccrine myoepithelial cells rarely contain LBs. The apocrine myoepithelial cells, in contrast, contain LBs in almost all cases. The apocrine secretory (luminal) cells never contain LBs, but do contain PASD-positive inclusions which can be misleading (Andrade et al., 2003) (figure 6).
A third gene (PRDM8) has recently been discovered in a single family. As with EPM2A and EPM2B mutations, the patients have a progressive myoclonus epilepsy which is similar to typical LD, but also exhibit significant differences (Turnbull et al., 2012). The sweat glands in these patients contain no LBs. At present, we have not been able to study the brain of these patients for lack of material. Absence of LBs in the sweat glands of these patients could influence the current diagnostic algorithm. If a patient has all the clinical manifestations of LD, a new gene defect is detected, and there are no LBs found in the sweat glands in the skin biopsy, the physician should consider an open muscle biopsy, as LBs are readily found in the muscle.
In order to further our knowledge of LD, it is necessary to study animal models of this disease which could lead to breakthroughs in the understanding of the pathogenesis, which may subsequently lead to potential therapies. Polyglucosan storage disease with myoclonic epilepsy has been known to exist in animals since it was reported in a beagle in the early 1920s. Although it has only been reported in several breeds of dogs and other species of the Canidae class, as well as cockatiels, pigs and cattle, it has probably been overlooked in many other species, since it is rare for animals to be subjected to post mortem examination, especially non-domesticated animals. The most widely studied of these naturally occurring animal models is the pure bred dog. As these animals are bred from a limited gene pool, the potential for consanguinity is greatly increased. Fortunately, breeders are regulated by kennel clubs and in order for these dogs to be sold as pure bred, evidence of a known lineage and a stringent medical examination must first be submitted to these organizations, prior to a certificate being issued. As the majority of these animals are sold as companion animals, the dogs are routinely examined by qualified veterinarians and sometimes, although rarely, LD is detected in these dogs. LD in dogs can occur spontaneously in any breed of dog, but it particularly affects miniature wire-haired dachshunds, basset hounds, and beagles (Kaiser et al., 1991; Gredal et al., 2003; Lohi et al., 2005). The disease does not present in dogs until at least five years age (35 in human years).
Myoclonus is the dominant feature of the canine disease and this can be induced by flashing lights, sudden sounds, and movement. Generalized or complex partial seizures can be seen in some dogs. The disease progresses over many years and gradually other neurological deficits, such as ataxia, blindness, and dementia, occur. Unlike humans, affected animals have a near-normal life expectancy, though as quality of life diminishes, the owner may be forced to euthanize the animal.
Pathological examination of these dogs revealed distribution and frequency of LBs in the brain identical to that found in the human disease (Lohi et al., 2005). As with humans, the liver contains LBs only in periportal hepatocytes. Both cardiac and skeletal myocytes contain LBs. Dogs do not have sweat glands in the skin, except in the footpads, where both apocrine and merocrine sweat glands are present; the LBs are identical in size and structure to those of the human disease. They are found in the apocrine myoepithelial cells and in the merocrine ductile and myoepithelial cells (figure 7).
With the identification of two genes (EPM2A [laforin] and EPM2B [malin]) responsible for LD, it was only logical that transgenic mice technology be utilized to develop murine models of the disease, in order to further advance our knowledge of LD.
The first of the animal models developed was a laforin-deficient murine mutant (Ganesh et al., 2002b). This was achieved by deleting the dual-specificity phosphatase domain of the EPM2A gene. These null mutants developed LBs in the brain, specifically in the hippocampus, cerebellum, cortex, and brainstem. Neuronal degeneration was observed in younger mice, but the same phenomenon was observed in age-matched wild-type littermates, which is suggestive of neuronal remodelling during development. The onset of LB accumulation occurred at two months. The highest number of perikaryal LBs was found in the molecular layer of the cerebral cortex and, as with humans, perikaryal LBs had two layers of various proportions; the outer layer being pale and the core basophilic. ‘Ground glass’ LBs were found throughout. Mild focal neuronal degenerative changes were detected primarily in the Purkinje cells of the cerebellum and were particularly present in the later stages of the disease. Ultrastructural examination of these tissues revealed prominent LBs in many of the neurons in all of the affected areas. As with dogs, mice have no sweat glands in the skin, except in the footpads where LBs were found. LBs were also found in skeletal and cardiac myocytes. No LBs were detected in the liver using either PASD staining or electron microscopy, although they were reported to be present using an antibody against polyglucosan bodies.
A transgenic mouse was generated which overexpressed myc-tagged inactivated laforin, in order to trap the substrate of laforin (Chan et al., 2004). Myc-laforin was expressed 150-fold higher than endogenous laforin. LBs were formed in the neuronal perikarya and dendrites, liver, sweat glands in the footpad, and skeletal and cardiac myocytes. In the liver, hepatocytes with LBs were found in discrete clusters throughout zones 2 and 3. Using an antibody against myc with immunperoxidase staining or immunogold labelling, we were able to determine both the cellular and subcellular distribution of myc-laforin. Particularly striking was the staining of the LBs in cerebellar Purkinje cell somas and dendrites and stellate neurons, as well as the presence of discrete punctate structures consistent with LBs throughout the molecular layer of the cortex, and in some hippocampal and cerebral neurons. Overall, however, LBs in the brain in this model were much lower in number than in the knockout mouse mentioned above.
In addition, a malin knockout mouse was developed. The malin exon was removed by in vivo recombination and an embryonic stem cell line (ESL) was created. The ESL was aggregated and a chimeric mouse was generated. These were bred into a C57B6 background and the malin null mouse line was established from the resultant heterozygous mice (Turnbull et al., 2010). These mice, like the laforin knockout mice, developed LBs in the brain at around two months. These null mutants developed LBs in the brain in the hippocampus, cerebellum, cortex, and brain stem. As was the case for the laforin-deficient mouse, the LBs consisted of two populations. The perikaryal LBs consisted of two layers; the outer layer being pale and mildly eosinophilic and the core basophilic. These were found primarily in the cell bodies of neurons in the cerebral cortex molecular layer. “Dust-like” LBs were found throughout the grey matter. LBs were also found in the sweat glands of the footpads and in skeletal muscle and cardiac myocytes. Unlike the laforin knockout or mutant laforin mice, malin null mice had LBs in some of the periportal hepatocytes. Not unlike the human or canine liver LBs, they occupied the centre of the cells, pressing the nuclei and the majority of cytoplasm to one side, as well as the periphery of the cell. Under the electron microscope, the LBs formed lakes which consisted of clusters of ‘glycogen-like’ particles and fibrillar material. Other groups have also generated malin knockout animals with similar results. Pathological sample images from the third mouse model mentioned above are presented in figures 8, 9 and 10.
One of the most exciting developments in the study of LD using transgenic mice is the use of double knockouts. In LD, glycogen synthase (GS) over-activity is considered one of the primary culprits for the formation of polyglucosans (Vilchez, 2007). It has been hypothesized that the reduction in GS activity might prevent polyglucosan formation. Laforin-deficient mice were bred with mice deficient in PTG, a protein involved in activating GS, resulting in LD mice lacking the GS-activating effect of PTG. The resultant double knockout (DKO) mice have almost no polyglucosan or neurodegeneration, and no seizures (Turnbull et al., 2011b). The development of this DKO mouse demonstrates the importance of using this approach to create strategies for therapeutic interventions which ultimately may result in a cure for this devastating disease. Due to the inability to use this approach in humans, results from this and knockouts of other glycolytic pathway constituents in other DKO mice are likely to reveal molecules which, when inactivated or eliminated, lead to the control or cure of LD. Once these molecules have been identified, the development of small molecule antagonists against GS, its activators, and potential GS up-regulators will be developed as potential therapies for human LD.
Pathogenesis of Lafora disease
Normal glycogen remains soluble in the cell due to its highly organized structure, whereas polygluosans precipitate in the cell due to disturbances in the structure of the carbohydrate, and aggregate into LB (Tagliabracci et al., 2008). LBs are very similar in morphology to polyglucosan bodies seen in glycogen-branching enzyme deficiency, differing only in their cellular location. In both LD and glycogen-branching enzyme deficiency, polyglucosans form in neurons. In LD, they are exclusively seen in the cell bodies and dendrites. In glycogen-branching enzyme deficiency, they are seen in axons. The two diseases have markedly dissimilar clinical symptoms, which is most likely to be due to this difference in polyglucosan localization. LD is a progressive myoclonus epilepsy, whereas glycogen-branching enzyme deficiency patients suffer from an adult-onset motor neuron disease, similar to that seen in amyotrophic lateral sclerosis (ALS) (Bruno et al., 1993).
EPM2A encodes a protein named laforin which contains a dual specificity phosphatase domain and a carbohydrate binding motif (Minassian et al., 1998). The second LD gene, EPM2B, encodes malin, an E3 ubiquitin ligase (Chan et al., 2004). Over a decade has passed since the second causative LD gene was identified. Since then, a number of hypotheses have been put forward as to the function of both malin and laforin in the pathogenesis of LD. The next decade will, most likely, unravel the complete pathway leading to LD and provide the knowledge which is necessary for finding a cure for this devastating disease.
LD has been speculated to be caused by defects in the clearance system(s) of the cell, similar to diseases such as Parkinson's and Alzheimer's. The presence of large inclusions and LBs, along with the finding that LBs contain a minor component of protein(s), suggested a defect in either autophagic processes and/or protein clearance. In keeping with this line of thought, defects in both autophagy and protein clearance have been reported in both Epm2a-/- and Epm2b-/- mice (Garyali et al., 2009; Aguado et al., 2010; Knecht et al., 2010; Puri & Ganesh, 2010; Rao et al., 2010; Criado et al., 2011; Puri et al., 2012). Unexpectedly, the autophagic dysfunction in Epm2b-/- mice is different to that of Epm2a-/- mice, resulting from an mTor-independent pathway (Criado et al., 2011). This suggests that if autophagic dysfunction is, indeed, at least partly causative of LD, mutations in malin and laforin may have separate and dissimilar disease mechanisms, which would be surprising, given the highly similar phenotypic outcomes in both laforin- and malin-deficient LD.
Malin has been reported to ubiquitinate a number of proteins, including laforin (Gentry et al., 2005), glycogen-debranching enzyme (Cheng et al., 2007), PTG/GS (Vilchez, 2007; Solaz-Fuster et al., 2008; Worby et al., 2008), neuronatin (Sharma et al., 2011), AMPK (Moreno et al., 2010), and dishevelled2 (Sharma et al., 2012). However, only laforin and GS have been clearly shown to be increased in Epm2b-/- mice (Tagliabracci et al., 2007, 2008; DePaoli-Roach et al., 2010; Turnbull et al., 2010; Valles-Ortega et al., 2011), indicative of in vivo action of malin on laforin. Additionally, no changes affecting the ubiquitin-proteasomal system were seen in Epm2b-/- mice (Criado, 2011), while these defects have been seen in a number of laforin-deficient models. It therefore appears that these protein clearance defects are a secondary consequence of LB formation, which is supported by recent studies showing that removal of the major polyglucosan component of LB by downregulation of glycogen synthesis results in near-complete disease resolution (Turnbull et al., 2011; Duran et al., 2014).
A major prevailing hypothesis over the years in LD research has focused on the balance between glycogen synthesis and glycogen branching (Lohi et al., 2006). Normally, GS synthesizes glycogen while the branching enzyme promotes the extension of the growing chain. This balance results in a soluble, highly ordered glycogen molecule. When this balance is disturbed in the direction of synthesis (i.e. when branching enzyme is deficient), polyglucosans form, as is seen in patients with glycogen-branching enzyme deficiency. The similarities between LBs and the accumulations seen in glycogen-branching enzyme deficiency, as well as data showing that over-expression of GS can also cause polyglucosan accumulation (Raben et al., 2001), make this an appealing and logical hypothesis. More substance was added to this proposal that there is a misbalance between synthesis and branching, when Fernández-Sánchez and colleagues demonstrated an interaction between laforin and PTG, a glycogen-targeting subunit of protein phosphatase-1 (PP1) (Fernandez-Sanchez et al., 2003). PTG directs PP1 to the glycogen-metabolizing enzymes, activating GS while inhibiting glycogen phosphorylase, and thus leading to a net increase in glycogen synthesis (Printen et al., 1997). In addition to its interaction with PTG, laforin has also been shown to interact with other regulatory subunits of PP1, including GL and R6, members of the same family as PTG. Subsequent studies have confirmed the PTG interaction using cell over-expression systems in vitro, but no in vivo interaction studies have been reported, which is likely to be due to a lack of a suitable antibody to PTG. Interestingly, some disease-causing mutations in laforin specifically affect its interaction with PTG, indicating that disruption of this interaction is pathogenic. Dysregulation of PTG has been hypothesized to cause LD, primarily based on results from in vitro studies. Elegant work by Vilchez and colleagues showed that malin and laforin act together to control levels of glycogen synthesis by targeting PTG and/or GS for proteasomal degradation (Vilchez, 2007; Solar-Fuster, 2008; Worby et al., 2008). When either laforin or malin is missing, levels of PTG, an indirect activator of GS, increase, causing GS over-activation. In this hypothesis, over-active GS would cause an imbalance between glycogen branching and extension, leading to the formation of LBs. Results from studies using animal models of LD showed no increases in PTG levels or GS activity, indicating that dysregulation of PTG may not be causative of LD (DePaoli-Roach et al., 2010; Turnbull et al., 2010; Valles-Ortega et al., 2011). Nonetheless, the large amount of in vitro data, along with a study showing that removal of PTG from laforin-deficient mice resulted in a dramatic reduction in LB and a cure for LD in the mouse, strongly supports the conclusion that PTG and laforin interact, and that both are involved in the same metabolic pathway.
The beginnings of an attractive hypothesis emerged from the laboratory of Jack Dixon when they reported that laforin was a carbohydrate phosphatase which could dephosphorylate amylopectin, a plant carbohydrate (Worby et al., 2006). Following this, Tagliabracci and colleagues expanded on this work in a series of elegant experiments. First, they demonstrated that laforin could indeed dephosphorylate glycogen and, most importantly, that glycogen phosphate levels were present at a much higher level than normal in mice lacking laforin (Tagliabracci et al., 2007, 2008). The covalently bound phosphate on glycogen was shown to be present at all available glucose carbons of glycogen, i.e. C2, C3 and C6 (Tagliabracci et al., 2011; Nitschke et al., 2013). Precisely where the phosphate on glycogen originates and how it results in glycogen becoming polyglucosan remains unknown. It was shown that laforin removes these phosphates during glycogen degradation (Irmia, 2015). This emerging phosphate hypothesis is compelling, but fails to identify a role for malin in LD. At 12 months of age, laforin-deficient mice have a 4.2-fold increase in muscle glycogen phosphate, whereas malin-deficient mice present a more modest increase of 2.8-fold over normal wild-type levels (Tagliabracci et al., 2008; Tiberia et al., 2012). The lack of a comparable phosphate increase in malin deficiency when laforin deficiency and malin deficiency are clinically equivalent raises the likelihood of other mechanisms at play in malin-deficient LD. Based on our most recent results from malin-deficient mice, we showed that absence of malin leads to increased laforin, as an initial step prior to any LB formation, and then progressive accumulation of laforin in glycogen. We also found that the gradual accumulation of laforin in glycogen renders the latter progressively less soluble. In related work, we showed that over-expressing laforin in cell culture leads to a conversion of glycogen to polyglucosan masses. This phenomenon continues to occur when laforin mutants, which bind glycogen but are phosphatase-inactive, are used. This no longer occurs when laforin mutants which cannot bind glycogen are used. Collectively, these results suggest that malin functions to regulate the quantity of laforin, and excess laforin on glycogen is detrimental to glycogen, leading to glycogen conversion to polyglucosan and LB (Tiberia et al., 2012).
The last decade has brought about a number of breakthroughs in the understanding of LD pathogenesis, though a clear picture has not yet formed. Each new hypothesis has come with its own weaknesses. The two main hypotheses of LB formation centre round the synthesis of glycogen and its structure. Compelling evidence has been presented for both, though each has their own caveats. Tiberia and colleagues attempted to propose a unifying hypothesis (Tiberia et al., 2012). Firstly, they considered laforin's role as a glycogen phosphatase, which removes phosphate from glycogen allowing it to remain soluble in the cell. Secondly, malin acts on laforin, and is likely also to act on other glycogen metabolic enzymes, to remove them from the glycogen molecule. If laforin is missing, phosphate accumulates and causes glycogen to become structurally abnormal and precipitate. If malin is missing, laforin remains “stuck” to glycogen, again disturbing the precise structure of glycogen and causing it to precipitate. This hypothesis answers a number of outstanding questions in LD research, foremost addressing the role of malin in LD. Here, malin has two roles; the first to control cytosolic amounts of laforin to keep it from inadvertently ‘clogging up’ the structure of glycogen, and second, to remove laforin (and probably other proteins, such as GS) from glycogen itself after laforin has removed the specific phosphate. Gentry and colleagues (Gentry et al., 2005) had previously speculated that malin may play a role in the regulation of laforin levels, but the actual mechanism behind this was unknown. In this hypothesis, both laforin deficiency and malin deficiency lead to LB formation by means of foreign matter on the glycogen molecule itself (phosphate and protein, respectively). While this theory appeared to bring together disparate aspects of LD research, most recent results once again raised a new and serious challenge. It was shown that expressing a phosphatase-inactive laforin in laforin knockout mice can fully rescue LD in these mice (Gayarre et al., 2014). This finding puts back into question the role of phosphate in polyglucosan formation, causing the field to be re-examined once more. The field has, however, been very rich in results since the genes for this disease were identified. It is hoped that a few new breakthroughs will lead to a comprehensive understanding of this deadly disease.
Disease management and therapy
Lafora disease (LD) is a devastating condition, with severely limited life expectancy and therapeutic options which continue to be limited. Its striking clinical and EEG features may lead to early diagnosis, but because it remains uncommon, few clinicians gain significant experience of this condition, and diagnosis is often delayed. Moreover, LD is a recessively inherited condition, with founder effects and increased prevalence associated with inbreeding, and thus LD is unevenly distributed across the world, which contributes to ‘knowledge gaps’ about the condition.
We will try here to delineate the practical steps that we recommend in the management of patients with LD. It is clear, in our eyes, that there is an intermediary stage: the clinical diagnosis of LD is now comparatively easy to confirm. Efficient anticonvulsants can be used to alleviate the burden of the attacks, but better insights into the mechanisms underlying the production of LBs and the progression of clinical symptoms have not yet produced ground-breaking therapeutic advances. Thus, dealing with LD patients and their families is not an easy task, because there is little hope to offer and the genetic nature of the condition raises many questions within the affected families.
Management: diagnosis and genetic counselling
Given the severity of the prognosis, a biological confirmation of the diagnosis of LD is necessary; this used to be made based on typical pathological findings (from a skin, muscle, or liver biopsy), but nowadays relies on genetics. Genetic screening, which remains costly, should be justified by solid clinical and EEG evidence, and should be performed together with a variety of tests, while screening for all possible genetic mechanisms in a poorly-assessed case with epilepsy and myoclonus.
The diagnosis of LD is based on three levels of evidence (figure 11):
- –The clinical evidence is a compound of history-taking (family background, circumstances, aspects and progression of seizures and myoclonus, and visual agnosia) and examination (including cognitive and psychological assessment, exclusion of associated symptoms such as sensory deficits, and video documentation of myoclonus and general behaviour). The most common clinical situation is one where idiopathic generalized epilepsy (IGE) or even juvenile myoclonic epilepsy (JME) has been diagnosed, with a re-assessment of the patient's situation because of a very unusual evolution which tends towards worsening; the clinical work-up should help exclude the possibility of aggravation of IGE by inappropriate antiepileptic drugs (AEDs), which may result in a pseudo-PME.
- –Complementary evidence is based on a thorough evaluation of the EEG, polygraphic EEG, and video-EEG (with an assessment of progression of changes over time). Neurophysiology may also help distinguish between LD and other adolescent-onset PMEs, e.g. Unverricht-Lundborg disease, in which the EEG changes are less pronounced, or juvenile ceroid-lipofuscinosis, in which prominent single-flash responses on the EEG are found. Other procedures may also help in the differential diagnosis (neuroimaging/MRI is not informative in LD). Pathological demonstration of LBs in sweat gland duct cells on axillary skin biopsy (or, less commonly, on liver or muscle biopsy) used to be the definitive diagnostic tool, but requires a highly trained pathologist; it may still render services as a supplementary element in favour of a diagnosis of LD when genetic testing is not available, or not entirely conclusive.
- –The confirmation of diagnosis is nowadays provided by the demonstration of a pathogenic mutation in both alleles of one of the EPM2 genes, with presence of heterozygous mutations in each of the clinically unaffected parents.
The diagnosis of LD can be made, or suspected, in various clinical settings, with, in our opinion, three typical situations:
At an advanced stage of the condition, in very typical patients and/or families who were not diagnosed earlier due to a lack of local expertise resulting from the rarity of LD in some settings, or due to the absence of molecular biological tools, e.g. in rural Africa (Traoré et al., 2009). In such cases, genetic testing will show the genetic subtype, and contribute to the worldwide ‘map’ of mutations found in EPM2A and EPM2B.
A PME or LD should enter the differential diagnosis for any adolescent with a diagnosis of IGE, JME, or photosensitive epilepsy with not only failure to respond to AEDs, but also a condition that appears to worsen. In such patients, a thorough re-evaluation of the clinical data shows that the patient has resting and action myoclonus, often well-controlled/masked by medication, such as valproate (VPA), or, characteristically, negative myoclonus (Genton et al., 2012), visual agnosia, and prominent, specific EEG changes. The search for a mutation in the known LD genes is warranted to confirm the diagnosis.
In other radically different situations within the context of a family with a confirmed diagnosis of LD in one of its members. Although LD is highly unlikely in asymptomatic adults, such adults will want to know whether they carry a heterozygous mutation; in younger asymptomatic siblings, LD may still manifest, and the emergence of “preventive” treatment strategies may render the identification of the mutation useful in this context. As the pathogenic mutations have already been characterized in the proband, the cost of the investigations for other family members will be lower, and the importance of detecting preclinical cases will also be known.
It is evident that the diagnosis of LD should not be given to the family before a final and definitive confirmation, however, once it is confirmed, it should not be kept from the caregivers. Presenting the diagnosis to patients themselves is not advisable, as there is little hope to offer. Moreover, they may be too young or affected to understand the implications; at an early stage, some may still be able to understand and gain access, e.g. on the internet, to detailed information on LD, which might cause severe depression. In our experience, a diagnosis of ‘severe, drug-resistant epilepsy’ will satisfy the patient's curiosity and still offer some hope and a reason for accepting help. However, if significant progress occurs in the near future, this global attitude may have to change, for example, in order to accept pathogenetically-founded therapies.
The consequences of a diagnosis of LD for the family of the proband should not be underestimated. Time should be devoted to the change expected for patients and their families (see below), but also to counselling regarding the following topics:
- –The legitimate feelings of guilt and resentment must be alleviated with a few simple statements, that the inheritance is bilateral (i.e. the disease does not come specifically from the father or from the mother, but from both sides), that the disease results from a very uncommon co-occurrence of abnormal genes (even in consanguineous marriages), and that persons with a single abnormal gene will not have the disease.
- –The main concern is the possible occurrence of LD in other family members. The risk can be practically excluded in older, fully asymptomatic siblings, but cannot be excluded in younger asymptomatic siblings nor, a fortiori, in as yet unborn siblings, if the parents are young enough to have other children. Whether siblings, especially younger ones, should be subject to molecular screening is still debatable. There is no reliable, recommended treatment, in pre-symptomatic LD cases, to prevent or delay the appearance and progression of symptoms. However, new perspectives are opening up with new possible treatments. Knowledge of the type of causative mutation that affects the proband allows precise counselling in this matter. Having provided all the relevant information, the clinician in charge of the patient will come to an agreement with the family and obtain their informed consent for all the procedures performed on non-affected family members. If the family shows reluctance towards genetic testing, a simple EEG recording for a younger sibling may provide information on the potential risk of LD, as EEG changes may precede the first clinical symptoms by many years (Van Heycoptenhamm & De Jager, 1963). Concerning future pregnancies for the parents of a patient with LD, the risks are known (LD: ¼; transmission of one pathogenic gene: ½; no risk: ¼), and prenatal screening can also be proposed.
- –The risk of carrying an abnormal gene and transmitting the condition to other generations is also a major concern in families with LD. We have no experience of patients with LD having children. Their siblings or parents (when they plan to have children with another spouse), as well as other family members, may benefit from molecular screening in order to assess the presence or absence of the pathogenic gene(s) found in the proband.
An important aspect of diagnosis and genetic counselling is financial; insurance and conditions of reimbursement (or, more basically, the availability for procedures, tests, and medications) differ greatly between countries and social systems. Families should always be informed about the costs involved; searching for a mutation is expensive, but the simple screening for a known mutation is less costly. Similarly, the regulations covering genetic diagnosis, screening, and counselling may differ between countries, and clinicians should always conform to local laws. Obtaining informed consent for the successive steps of diagnostic and screening procedures is a minimum requirement.
Management: treatment and social support
In this article, new therapeutic proposals are not discussed since these are covered in detail in the last article (Minassian, 2016). We shall focus on the medical and social measures that are available for the care of present-day LD patients (figure 12). It is clear that health systems vary greatly between countries, particularly between affluent and less developed societies. Moreover, in many developed countries, health coverage is not equal; some patients (e.g. public sector employees and their families) may benefit from a whole range of possibilities, while others (e.g. those informally employed in the private sector) do not. It is also clear that patients with LD should benefit from the maximum possible help provided by the available health system. LD is a rare condition which cannot be considered as a public health problem (as costs are limited by the small number of affected persons), but it has major consequences for patients and caregivers, and society should show solidarity and provide all available support.
With regards to treatment, AEDs only have a partial effect on myoclonus and seizures, and there are none that have a major influence on the progression of cognitive and behavioral symptoms. Patients typically receive an AED, usually valproic acid, after the first generalized tonic-clonic seizure (GTCS). This is usually effective in suppressing, for some time, most GTCS, the symptoms associated with photic sensitivity, and some of the myoclonus. There are two unusual effects, which should lead to an early diagnosis of LD: first, the EEG shows rapidly increasing, permanent interictal changes, including focal occipital spikes, despite the apparent clinical remission; second, the patients develop negative myoclonus, which becomes prominent before the more characteristic myoclonic jerks. Other AEDs are used during this stage: lamotrigine (LTG) is not very advisable in the context of a myoclonic epilepsy, but may help transiently; phenobarbital (PB) and primidone (PRM) are effective, but are often used at high doses and their cognitive effects are added to those of the condition; and levetiracetam (LEV) is increasingly used early for adolescents with IGE, hence in LD cases, even before confirmation of the diagnosis. Other helpful drugs include topiramate (TPM) and zonisamide (ZNS), which both have marked antimyoclonic effects in some patients. Additional relief can be obtained, often transiently, with ethosuximide, felbamate, methsuximide, and benzodiazepines (BZD). The latter (usually clobazam, clonazepam, and diazepam) should be used with care since there is a marked initial effect followed by quick tolerance. Finally, there have been two recent single case reports of rather dramatic beneficial effects of perampanel (Schorlemmer et al., 2013; Dirani et al., 2014), and a larger study is presently underway.
With such a severe condition, the paradoxical aggravating effect of some AEDs may be difficult to pinpoint. There is no evidence that carbamazepine (CBZ), oxcarbazepine (OXC), phenytoin (PHT), eslicarbazepine, gabapentin, pregabalin, vigabatrin, or lacosamide are of any benefit. Often, withdrawal of one of these AEDs (especially CBZ or OXC) will bring some relief. However, we have experienced cases in which status epilepticus responds well to phenytoin loading. Phenytoin should not, however, be kept as maintenance medication subsequent to arresting the status.
With the progression of LD, AED treatment progresses to polytherapy, with a combination of several of the drugs quoted above (with the exclusion of LTG); the commonly used combinations are VPA+TPM or ZNS or LEV, with an additional BZD, a 3- to 5-drug combination being fairly common; one can switch between BZD when tolerance occurs. In case of severe, transient aggravation, with serial seizures or status epilepticus, there should be no abrupt changes to the usual regimen (except for the interruption of a potentially aggravating AED), and IV BZD should be used, as well as, for a limited period, IV PB or PHT. In our recent experience, the final progression of the disease no longer includes refractory status epilepticus, but more commonly involves non-specific complications, infectious or otherwise, in bedridden and demented patients. Thus, despite their lack of influence on the overall evolution of the disease, modern AEDs have partly changed the outcome, which is apparently no longer accompanied by severe, formerly often terminal, episodes of refractory convulsive status epilepticus.
In LD, social support is at least as important as medical treatment. Psychological support can be provided by patient organisations and there are several which are specifically devoted to LD (table 2). Individual patients should also receive professional psychological support during the early stages of the condition. Physical therapy aims at maintaining a good overall muscular condition and at preserving ambulation, for as long as possible.
At the onset of seizures, patients are usually in secondary school and experiencing increasing difficulties with academic requirements. In order to enable them to maintain social contacts, it is best to maintain schooling for as long as possible, while negotiating with teachers about academic performance. However, this cannot be kept up for extensive periods. Some families will choose to keep patients at home with the best possible environment; this often requires adaptive measures, avoiding the use of stairs, setting up the patient close to the bathroom, and providing 24-hour presence at home with the help of a health professional to check medications. Other families will seek a specialized institution where patients are kept with other epilepsy patients in the same age group, with some amount of education and social activities. Re-evaluation at the specialized neurological department can be organized at 6- or 12-month intervals, with acute admissions in the event of complications, often due to intercurrent diseases (e.g. febrile infections) and/or worsening of epilepsy.
The latter period is characterized by increasing dependency as the patient becomes wheelchair bound and, later, bedridden. According to local availability and the wishes and capacities of the caregivers, the patient is maintained at home, institutionalized or hospitalized. There should always be a connection between the reference specialized epilepsy team and the local caregiving structure.
Conclusion
For patients with LD, the present state of possibilities includes a logical, effective approach to diagnosis, and the rational use of all available tools to help both the patients and their families. We hope that the management of LD patients will undergo a profound change in the near future, when effective, pathogenetically-oriented treatments become available.
Disclosures
None of the authors have any conflict of interest to disclose.
The present manuscript is part of a Epileptic Disorders/Mariani Foundation supplement on Progressive Myoclonus Epilepsies, downloadable as a whole by visiting www.epilepticdisorders.com.