Epileptic Disorders
MENUKCNQ2 mutation in an infant with encephalopathy of infancy with migrating focal seizures Volume 20, issue 6, December 2018

Figures
- Key words: epileptic encephalopathy, KCNQ2, migrating focal seizures, potassium channels, neonatal seizures
- DOI : 10.1684/epd.2018.1011
- Page(s) : 541-4
- Published in: 2018
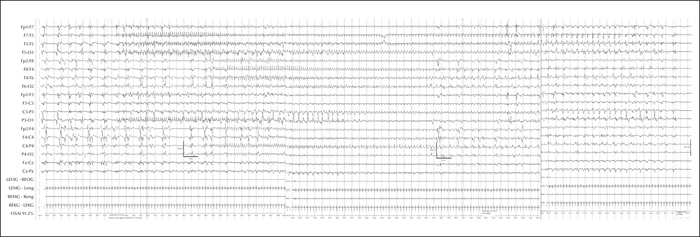
A male neonate presented with seizures at 18 hours of life, characterized by tonic posturing with eye deviation to the right, apnoea, bradycardia, and oxygen desaturation. Initial structural, metabolic, and infectious work-up was unremarkable. He continued to have seizures refractory to a variety of antiepileptic medications. A phenobarbital coma was trialled, leading to cessation of clinical seizures but continuation of electrographic status epilepticus. On EEG, ictal discharges originated from both the right and left hemispheres, migrating to the opposite hemisphere, consistent with encephalopathy of infancy with migrating focal seizures. At this time, he developed septic shock and was trialled on a ketamine infusion and ketogenic diet. Due to his poor prognosis, a goals of care discussion was carried out with the family, leading to withdrawal of care and his subsequent death at one month and seven days. A posthumous genetic panel revealed a de novo KCNQ2 p.Ser247Leu variant, considered likely to be pathogenic. This is the third reported case of a KCNQ2 mutation associated with an encephalopathy of infancy with migrating focal seizures phenotype. We discuss potential cellular mechanisms underlying this unique KCNQ2 phenotype, as well as future therapeutic considerations.