Epileptic Disorders
MENUJuvenile myoclonic epilepsy phenotype in a family with Unverricht-Lundborg disease Volume 21, issue 4, August 2019

Authors

1 Razi Hospital, Department of Neurology, LR 18SP03, Tunis
2 Université de Tunis El Manar, Faculté de Médecine de Tunis, Tunis, Tunisia
3 Institut national de la santé et de la recherche médicale (INSERM), U975, ICM, Hôpital Pitié-Salpêtrière, Paris
4 Sorbonne Universités, Institut du Cerveau et de la Moelle épinière, ICM, Inserm U1127, CNRS UMR 7225, Paris
5 Institut du Cerveau et de la Moelle épinière, plateforme de génotypage et de séquençage, Hôpital Pitié-Salpêtrière, Paris
6 APHP, Hôpital Pitié-Salpêtrière, Département de Génétique, Paris, France
* Correspondence: Riadh Gouider
CHU Razi, Service de neurologie,
1, rue des Orangers,
Tunis, Tunisia
- Key words: Unverricht-Lundborg disease, juvenile myoclonic epilepsy, cystatin B, cathepsin B, cystatin C
- DOI : 10.1684/epd.2019.1078
- Page(s) : 359-65
- Published in: 2019
Aims
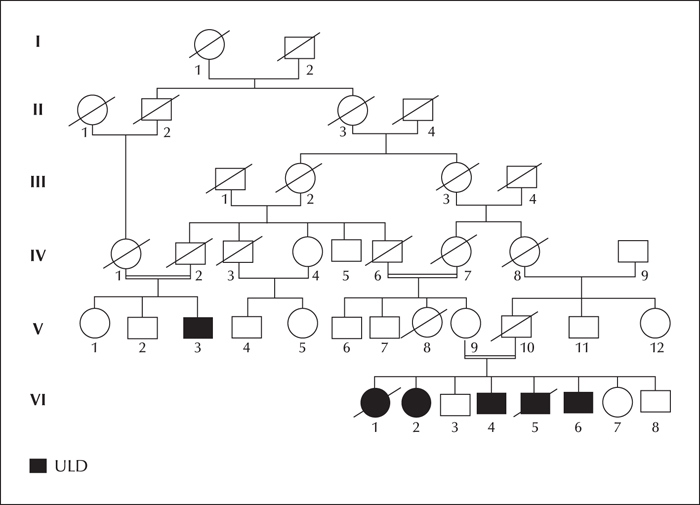
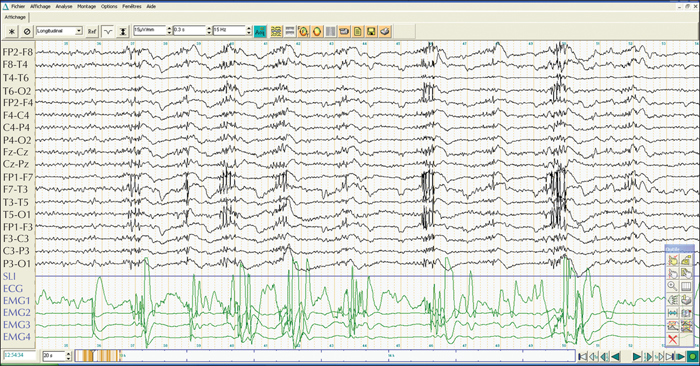
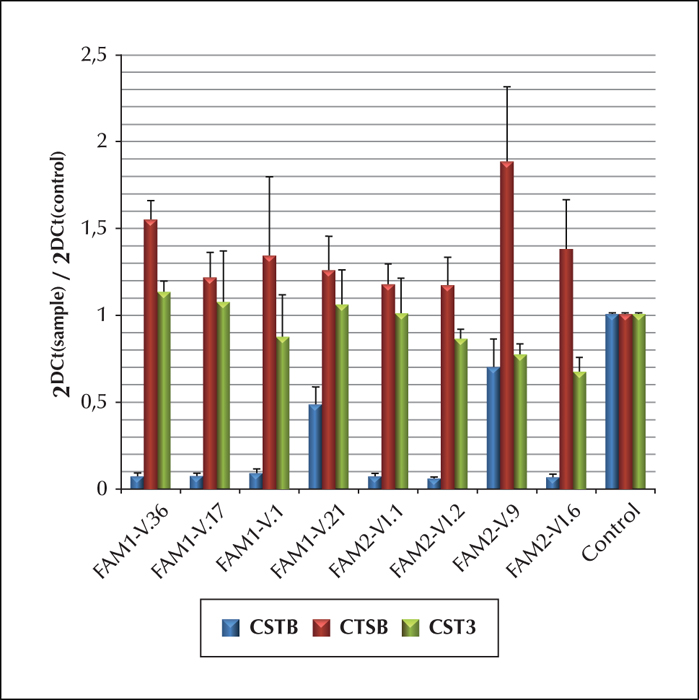
Unverricht-Lundborg disease (ULD), an autosomal recessive progressive myoclonus epilepsy, is due to an expansion, or less commonly a mutation, of the cystatin B (CSTB) gene. We report a clinical and molecular study of a Tunisian ULD family with five affected members presenting with a juvenile myoclonic epilepsy (JME)-like phenotype.