Epileptic Disorders
MENUEpilepsy in neurodegenerative diseases Volume 24, issue 2, April 2022

Professional expertise: Level 2: Proficiency and Level 3: Advanced Competencies and learning objectives:ILAE Curriculum
Neurodegenerative disorders are a group of conditions characterized by selective neuronal loss and by a progressive course and chronic evolution that lead to a gradual deterioration of functions. The spectrum of neurodegenerative diseases encompasses a large number of different entities with variable epidemiology, clinical manifestations, neuropathology and management [1].
The incidence of neurodegenerative disorders varies across different age groups with two age peaks in children/young adults and in the elderly. The most common causes of degenerative diseases with onset in the first two/three decades of life are genetic disorders, most of them producing neuronal damage or neurotransmitter alterations. In the elderly, age is the major risk factor associated with neurodegenerative conditions, such as Alzheimer or Parkinson diseases, vascular dementia or other dementia conditions of old age [2].
Epilepsy can be a comorbidity in a large variety of neurological disorders including neurodegenerative diseases [3]. Although the individual risk of epilepsy can vary significantly in the different neurodegenerative conditions, the possibility of a comorbid epilepsy has to be considered when evaluating a patient suffering from these disorders and presenting with episodes of possible epileptic nature, since the potential benefits from early diagnosis are high. Indeed, a prompt diagnosis in younger subjects can help achieve better cognitive development and quality of life outcomes, whereas in older patients, a better seizure control can minimize cognitive deficits and prevent morbidity from falls or other seizure-related injuries, in addition to enabling medical and legal assistance for complex decision making in their advanced ages.
Older age represents an independent risk factor in neurodegenerative diseases [4] and epilepsy [5]. Epilepsy occurs in about 1% of patients aged more than 65 years (about one quarter of newly diagnosed epilepsies) [2, 6]. In this age group, the most common pathologies underlying seizures are cerebrovascular diseases, brain tumours, traumatic brain injury and neurodegenerative disorders [4]. For these latter conditions, epidemiologic data, although scarce, indicate that they might account for about 10% of late-onset epilepsies [2]. Due to the lack of anatomical or biochemical markers for the neurodegenerative diseases at their onset, the recognition of a causal relationship between epilepsy and a degenerative condition is possible only in retrospect, when this latter disease becomes clinically evident. It is thus possible that a proportion of late-onset epilepsies classified as cryptogenic are instead related to neurodegenerative pathologies [7], suggesting also that the 10% figure emerging from studies mentioned above might underestimate the etiologic role of degenerative pathologies in epilepsy in the elderly. This consideration may advocate the inclusion of a comprehensive cognitive assessment for elderly patients with an initial seizure and negative etiologic investigation to yield an early diagnosis.
In this paper, we present an updated overview of the clinical features, diagnostic procedures and pathogenetic mechanisms of epilepsy in the most common degenerative diseases, aiming to provide a tool that can help epileptologists and neurologists in the diagnosis and management of this increasingly reported comorbidity.
Neurodegenerative disorders and epilepsy: clinical features, diagnostic procedures, and pathogenetic mechanisms
Alzheimer disease
Alzheimer disease (AD) accounts for 60-70% of all dementia cases [8]. The accumulation of extracellular aggregates of amyloid β (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) made of hyperphosphorylated tau-protein in cortical and limbic areas of the human brain is considered to play a major role in the neurodegenerative processes occurring in the brain of AD patients, as proposed by the amyloid hypothesis [9], even though other complex and multiple factors have been recently shown to participate in the development of dementia [10]. The first description of epileptic seizures in a confirmed AD patient is ascribed to Hannah in 1936 [11] although, in 1911, Alzheimer himself had already reported a patient with probable seizures and with amyloid deposition in the brain as the only pathological marker [12]. It is now widely accepted that seizures can occur in AD patients [13] and that AD-related pathological changes might be a causative factor for late-onset unprovoked seizures [14].
Epidemiology and modifying risk factors for seizures
The lifetime prevalence of seizures in AD patients varies in different studies depending on the sampled population or on whether they are retrospective or prospective. Although some reviews report a prevalence ranging from 10% to 22% [13, 15], the accuracy of these studies may be limited by the variability of the inclusion criteria that might have led to the inclusion of patients with other forms of dementia or other symptomatic causes of epilepsy in the elderly, besides pathologically confirmed AD [16].
The probability of developing seizures after AD onset has been estimated to be 13.4% [17], while a study on mild AD patients with a follow-up of seven years reported a cumulative incidence of unprovoked seizures of 8% [18]. Compared to healthy age-matched individuals, patients with sporadic AD have a two to ten-fold increased risk of manifesting seizures during the course of their illness [19]. Recently, a study analysing data from the Framingham Heart Study (FHS) showed a two-fold increase in risk for dementia among prevalent cases of epilepsy compared to controls, and a similar increase in risk of subsequent epilepsy among people with diagnosed dementia [20]. Similarly, the Atherosclerosis Risk in Communities (ARIC) study reported a three-fold increase in risk for new-onset epilepsy among people with dementia as well as a three-fold elevated risk of developing dementia in patients with late-onset epilepsy [21].
An increased risk of developing seizures has been observed in patients with younger age at AD onset [18]. About 15% of patients with early-onset AD have mutations in presenilin-1 (PSEN1), presenilin-2 (PSEN2), or amyloid protein precursor (APP) genes [22]. Around 20% of PSEN1 pathogenic variants have been reported to be associated with seizures, prompting the proposal to recognize a PSEN-1-associated genetic epilepsy syndrome [23]. Moreover, seizures can occur in carriers with mutations in autosomal dominant AD genes even if they are cognitively asymptomatic [24]. However, the occurrence of seizures may be underestimated, since in clinical practice the detection of seizures and the description of their semiology is based largely on information reported by the patient him/herself or by the caregiver. The impairment of memory in patients with dementia or the difficulties experienced by a caregiver in distinguishing fluctuating behavioural manifestations, common in demented patients, from focal seizures, may significantly hinder the recognition of epilepsy as a comorbidity or the evaluation of its severity.
Several studies have shown that seizure prevalence in AD seems to increase with disease duration [13, 25]. The mean interval from diagnosis of AD to seizure onset is reported to range from 3.6 years [25] to 6.8 years [26]. However, a seizure onset early in the course of the disease, even in stages preceding the onset of cognitive decline, has been documented as well, probably reflecting the well-established concept that the neuropathological alterations of Alzheimer disease precede the onset of symptoms [27]. A clinically-based study on newly diagnosed probable-AD patients revealed a history of seizure disorder in 6.8% of them [28]. In 3.4% of cases, seizure onset was time-locked to the onset of cognitive decline, and no symptomatic or provoking factor for seizures other than AD was identified [28]. Based on retrospective studies reviewing the prevalence of adult-onset epilepsy in a large population of AD patients, seizure onset was reported, on average, 4.6 years before the detection of cognitive symptoms [29].
These pieces of evidence have led to a debate on the existence of an inaugural epilepsy syndrome in sporadic AD that might define an “epileptic variant” of AD [30]. At present, however, according to the IWG-2 criteria for the diagnosis of AD, early occurrence of seizures remains an exclusion criterion for typical AD [31].
Seizure semiology
Focal seizures with impaired awareness with or without secondary generalization are the most common seizure type reported in up to 90% of AD patients [13]. However, subtle non-convulsive seizures without overt clinical symptoms may likely pass unrecognized and thus be under-reported. A study designed to identify possible ictal symptoms/signs compatible with epileptic seizures in a population with AD and mild cognitive impairment [32] identified multiple seizure-related features, such as altered responsiveness, speech/behavioural arrest, oral automatism, olfactory/gustatory aura, focal motor phenomena, other sensory symptoms (including hallucination) and amnesic episodes on awakening, suggesting that the most common of these manifestations might easily not be reported by caregivers because these are considered a feature of underlying dementia.
Transient epileptic amnesia (TEA), sporadically reported in the early stages of AD, has been suggested as a possible cause of wandering behaviour in AD patients [33]. Non-convulsive status epilepticus, even if occasional and rare, has also been described [34].
EEG
EEG, and in particular longitudinal EEGs, are not part of the usual clinical work-up in dementia patients. Non-epileptiform abnormalities such as theta or delta slowing are common EEG features of AD patients [35], but little information exists about the prevalence and diagnostic value of epileptiform abnormalities. In a large study [36] on individuals with different types of dementia, epileptiform EEG discharges were found in only 3% of the patients, which is a rate similar to that of the general population. Only 10% of the AD patients with epileptiform activity on EEG developed seizures later on during the course of the disease [36]. However, in another investigation on 33 AD subjects without a history of seizures, subclinical epileptiform activity was detected in 42.4% of patients and 10.5% of controls [37]. Finally, a study with foramen ovale electrodes showed that clinically silent mesial temporal lobe seizures and epileptic spikes, predominating during sleep (thus possibly interfering with memory consolidation), can be detected early in the course of AD, in the absence of significant scalp EEG abnormalities [38], further suggesting that epileptic activity or seizures may be undetected in a proportion of AD patients. All these data cast some doubts on the value of the standard scalp EEG in diagnosing epilepsy in AD patients and emphasize the need for larger longitudinal EEG studies, perhaps using additional techniques, including foramen ovale recordings, to determine the EEG diagnostic value in clinical practice.
Pathophysiology of seizures in AD and impact on cognitive functions
The pathophysiological mechanisms underlying epilepsy in AD are still incompletely elucidated. Experimental studies have shown that mice with APP mutations exhibit not only high levels of amyloid-beta (Aβ) peptides in the brain and develop AD-like clinical and pathological abnormalities, but also have spontaneous non-convulsive seizure activity in cortical and hippocampal networks [39]. Moreover, video-EEG monitoring of the cortical and hippocampal activity in human amyloid precursor protein transgenic (hAPP) mice, without evidence of neuronal loss, showed abundant epileptiform activity, suggesting that the exposure to pathologically relevant levels of Aβ may be sufficient to elicit aberrant network synchronization, epileptiform activity and seizures, even in the absence of frank degeneration [40]. Epileptic discharges trigger inhibitory rescue responses in hippocampal circuits that may contrast Aβ-induced aberrant network activity but also interfere with normal neuronal functions required for memory formation and learning [41]. Therefore, cognitive decline and epilepsy can reciprocally influence one another in AD and may possibly share some common pathophysiological mechanisms, suggesting that treating epilepsy, or if possible preventing it, could be an important new approach to slow down cognitive decline in selected patients with Aβ pathology. Moreover, hyperphosphorylated tau aggregates and NFTs have been observed in several patients with epilepsy [42] and pathologic tau has been correlated with epilepsy in animal models [43].
Therapy
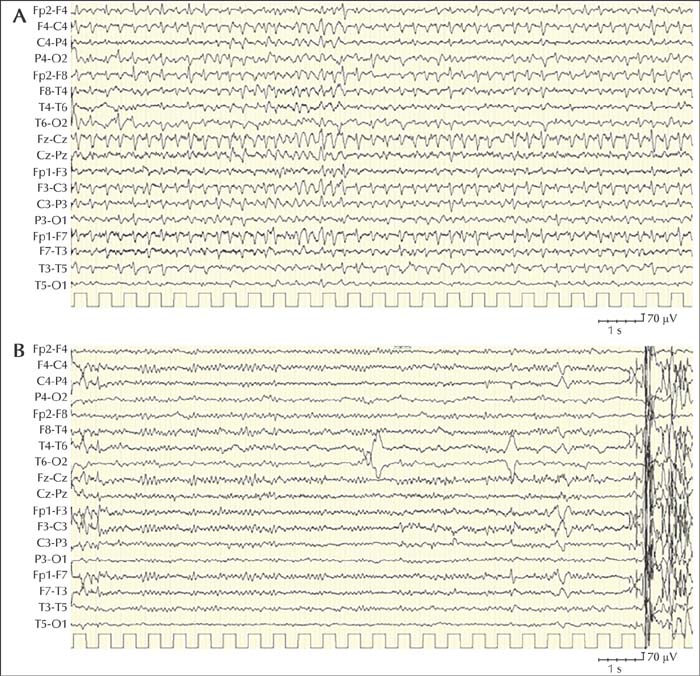
Antiseizure medications (ASMs) seem to prevent the recurrence of epileptic seizures in most people with Alzheimer disease, however, other factors such as drug-drug interactions, pharmacodynamics and adverse effects on cognition and behaviour should be taken into account for the treatment of seizures in AD patients. Long-term use of benzodiazepines is associated with an elevated risk of cognitive decline or dementia [44]. Valproic acid has been associated in clinical trials with a faster decline in Mini-mental state examination (MMSE) scores and more pronounced atrophy on brain imaging [45]. Lamotrigine, which is considered a viable option for treating seizures in patients with these [46], may exacerbate myoclonus in AD patients, especially those with PSEN1 mutations [23]. A recent Cochrane review investigating the efficacy and tolerability of pharmacological or non-pharmacological interventions for the treatment of epilepsy based on randomized and quasi-randomized controlled trials revealed no significant differences in seizure freedom for comparisons between levetiracetam versus lamotrigine, levetiracetam versus phenobarbital, or lamotrigine versus phenobarbital. Additional findings suggested that levetiracetam could improve cognition and lamotrigine could relieve depression, while phenobarbital and lamotrigine could worsen cognition, and levetiracetam and phenobarbital could worsen mood [47]. However, the level of evidence of these results was very low and the study failed to show significant differences in efficacy and tolerability between levetiracetam, phenobarbital and lamotrigine [47]. In conclusion, at present, there is insufficient evidence to support which ASM should be recommended to treat seizures in AD. An 89-year-old woman with chronic renal failure, diabetes and hypertension was diagnosed with Alzheimer's dementia at the age of 85 years. Neuropsychological evaluation showed deficits in executive functions, sustained attention and semantic and procedural memory; Mini-Mental State Examination score was 16 (normal range: 26-30). At the age of 89 years, she was admitted to the hospital because of an acute onset of “tremors” in the right upper limb, then spreading to the ipsilateral lower limb, without loss of consciousness, although she had difficulties in answering questions and executing simple orders. A CT scan and a biochemical work-up were unremarkable. During admission, the episodes of unilateral clonic jerking on the right side became progressively more frequent and consciousness was impaired which did not recover in the intervals between seizures. The EEG showed continuous spikes and spike-waves at the bifrontal leads, with left prevalence associated with myoclonus of the right limbs (figure 1A). A diagnosis of focal motor status epilepticus was made. The administration of 5 mg of midazolam IV was followed by EEG normalization (figure 1B), disappearance of the clonic jerks and recovery of consciousness. Chronic antiepileptic therapy with levetiracetam was undertaken. The patient was seizure-free at a six-month follow-up consultation. Case notes: This example illustrates a case of epilepsy comorbidity in AD. This patient presented with an isolated episode of focal motor status epilepticus that occurred four years after the onset of dementia, in agreement with data in the literature, which report an average interval of 3.6 years between the diagnosis of AD and the appearance of epileptic seizures. The focal status epilepticus was diagnosed based on the clinical features, the ictal EEG and electroclinical response to therapy. The patient started treatment with levetiracetam and she did not present further seizures during follow-up.Case 1
Down syndrome and late-onset epilepsy
Down syndrome (DS), the most common chromosomal disorder in humans, is due to trisomy of chromosome 21. Seizures or epilepsy, not reported in the first description of the disease [48], are known to have a higher prevalence (from one to 13%) than in the general population [49].
The onset of seizures follows a bimodal distribution, with 40% of DS patients presenting with epilepsy in infancy or early childhood, mainly with epileptic spasms and tonic seizures, and another 40% who start to suffer from epilepsy in the third decade of life [49], when Down patients start to be affected by a dementing process.
Autopsy studies demonstrated that neuropathological features of Alzheimer can be observed in the brain of DS patients from the age of 37 years [50], while the average age at onset of dementia is between 50 and 55 years [51]. According to a neuropathological hypothesis, triplication of the APP gene, located on chromosome 21, leads to amyloid overproduction. The extracellular accumulation of amyloid plaques induces synaptic degeneration, circuit remodelling and abnormal synchronization of neuronal networks, resulting in cortical irritability, thus with an epileptogenic mechanism similar to AD [52].
Accompanying or closely following the appearance of neurological deterioration, more than 50% of patients with DS and AD dementia will most likely develop epilepsy. The prevalence of untriggered seizures in DS increases dramatically after the age of 45 years, anticipating or in parallel with the emergence of cognitive deterioration. The prevalence of epilepsy can be as high as 46% in those over 50 years of age [53]. Myoclonic seizures are the most common seizure type, involving the limbs and the trunk. At onset, myoclonic seizures predominantly present in the morning, and polygraphic recordings show myoclonic bursts associated with generalized spike-wave discharges, as in juvenile myoclonic epilepsy [54]. Myoclonic seizures can occur in clusters, but massive and/or fall-inducing myoclonus are infrequent [54]. Generalized tonic-clonic seizures may appear at a later stage [55], as well as photoparoxysmal EEG responses [56]. Focal seizures with an altered level of consciousness without a motor component, the most frequent type of epileptic seizure in sporadic AD, occur rarely in DS [49]. This epileptic disorder, with predominant myoclonic features occurring in DS patients with dementia, has been proposed to represent a specific entity, labelled as “late-onset myoclonic epilepsy in DS” (LOMEDS) [57]. LOMEDS appears, on average, 6.9 months after the onset of dementia and exhibits a clinical course similar to that of progressive myoclonus epilepsies, with myoclonus becoming more severe over time and resistant to treatment, in parallel with progressive cognitive decline [56]. The development of multifocal epileptic and non-epileptic (i.e., action) myoclonus, sometimes induced by movements or less frequently by sensory stimulus (i.e., reflex epileptic myoclonus), increases the risk of falls. In the more advanced stages, myoclonia inconstantly display an EEG correlate [53], suggesting a non-cortical origin, and tremor and disabling cerebellar ataxia appear, worsening relentlessly [51].
The occurrence of epileptic seizures in DS has been associated with a more rapid cognitive and functional decline [51]. Epilepsy is also an independent risk factor for more frequent hospital admissions and mortality in adults with DS [53]. A study showed valproate and levetiracetam to be effective in controlling myoclonic and generalized tonic-clonic seizures in about 80% of subjects [54]. An overview of published cases with LOMEDS is presented in table 1.
Prion diseases
Prionopathies are a group of rare, fatal neurodegenerative diseases characterized by progressive dementia and focal neurological signs. The pathogenic hallmark of these conditions is the aggregation and accumulation of an abnormally folded protein termed PrPsc, which differs from the normal protein (PrPc) due to a high number of β-pleated sheets in the secondary structure compared to normal α-helices. This conformation renders the protein capable of resisting protease degradation. The prion protein can also determine the conversion of normal PrPc to PrPSc, causing an accumulation in certain populations in neurons, leading to vacuolation, astrocytosis, development of amyloid plaques and neuronal loss. Sporadic, genetic and acquired forms of prion diseases have been described [58].
Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common prion disease, accounting for about 85% of CJD cases, with an incidence of one to two cases per one million population per year [59]. Autosomal dominant CJD is the next most common, caused by mutations in the PNRP gene, encoding for the prion protein. The rarest forms are those that are acquired, such as kuru, iatrogenic CJD and variant CJD (vCJD) [58].
Current data on epilepsy in prion diseases are based on small series or individual case reports, in which epilepsy is often superficially described. Overall, epilepsy appears to be a very rare comorbidity in prionopathies. In sCJD, seizures can occur during the course of the disease in about 8% of subjects [3]. Seizures, as the presenting symptom of sCJD, are even rarer, occurring in only about 3% of cases [60].
The inherited forms of the disease, caused by mutations of the PRNP gene at chromosome 20, include Gerstmann-Straussler-Scheinker disease (GSS), fatal familial insomnia (FFI) and familial CJD. For familial CJD, the most common mutation is the E200K point mutation. In this latter type of CJD, seizures have been reported to occur in up to 40% of individuals [61].
Iatrogenic cases are caused by neurosurgical instruments, implantation of stereotactic electroencephalogram electrodes or by transplanted human tissues (dura mater grafts, corneas), contaminated by PrPsc protein. In contrast to what has been observed in sporadic and genetic cases, in iatrogenic CJD, seizures are exceptional or completely absent [62].
To our knowledge, no reports of seizures exist in the acquired form of prion disease, kuru, while focal motor seizures have been described in only one case with variant CJD [63].
In patients with prionopathies and epilepsy, seizure onset may precede the onset of typical symptoms by a few weeks [64]. However, the recognition of epilepsy as a presenting symptom may be overlooked, due to the difficulties in distinguishing, at disease onset, dyscognitive or psychiatric symptoms from subtle non-motor seizures with impaired awareness. In almost half of the patients reported in the literature, non-convulsive status epilepticus is the most frequently reported type of seizure, followed by focal aware seizures with motor phenomena (often myoclonic jerks of the limbs), focal seizures with impaired awareness, focal motor status epilepticus (epilepsia partialis continua), convulsive status epilepticus and focal to bilateral tonic-clonic or generalized motor seizures.
The physiopathogenetic mechanisms underlying epilepsy in prion diseases are still poorly elucidated. Studies in vivo have demonstrated that deletion of the PrPc gene in mice enhances neuronal excitability and seizure sensitivity by disrupting Ca2+-activated K+ currents, generating abnormal GABA-A inhibition in the hippocampus and/or higher levels of neocortical and subcortical oxidative stress [65]. Loss of PrPc function [66] due to conversion to, or contact with PrPsc and toxic processes due to propagation of abnormal PrPsc may contribute to destabilize local circuits and generate hyperexcitable and synchronized epileptogenic networks.
The typical electroencephalographic findings in prion diseases are periodic sharp-wave complexes (PSWC, otherwise known as triphasic waves or TWs), which can be found in all types of prion diseases, but the new variant (vCJD) [67]. PSWCs are frequently bilaterally distributed but may appear, especially in the first stages of the disease, lateralized, resembling periodic lateralized epileptiform discharges (PLEDs) [68]. PLEDs have been described in CJD patients with focal motor seizures [69], focal motor status epilepticus [70] and focal non-convulsive status epilepticus [71]. Different criteria for diagnosing seizures in CJD have been used, such as correlating the EEG patterns with the clinical manifestations (i.e. in focal motor seizures) [70] or altered mental status [72], assessing the electroclinical improvement after ASM administration or evaluating the evolution and variability of EEG patterns during prolonged recordings [72]. However, the reliability of EEG recordings in diagnosing seizures in CJD is still debated [73], especially to discriminate between non-convulsive status epilepticus (NCSE) and the typical alterations of consciousness observed in the late stages of the disease, since a misdiagnosis of refractory non-convulsive status epilepticus can lead to an aggressive but unnecessary therapeutic approach. Indeed, it is still discussed whether, in CJD, EEG abnormalities suggestive of NCSE reflect true seizure activity or whether they are PSWCs. Higher frequency (2.4 Hz vs 1.8 Hz) of the epileptiform activity and clinical-EEG improvement after benzodiazepine administration have been considered to be compatible with NCSE. In some instances, serial or prolonged EEGs can be necessary to establish the correct diagnosis [73].
In polygraphic and video EEG recordings of 109 sCJD patients [74], myoclonic jerks were observed in 50% of the subjects. PSWCs were detected in all but one patient with myoclonic jerks, and they were time-locked to the EMG myoclonic bursts only in the case of periodic myoclonus, supporting studies that demonstrated primarily a subcortical origin of myoclonus in the terminal stage of CJD.
Anecdotical neuroimaging studies in patients with sporadic and variant CJD, besides the MRI findings that characterize CJD (such as abnormal diffusion in the cortex, caudate, and/or putamen and abnormal FLAIR cortical hyperintensities), have shown alterations compatible with prolonged seizures or status epilepticus as well as peri-ictal changes in the cerebral cortex, hippocampi and thalamus (particularly the pulvinar region), or cortical and gyriform diffusion signal hyperintensity with cortical ribbon oedema [75].
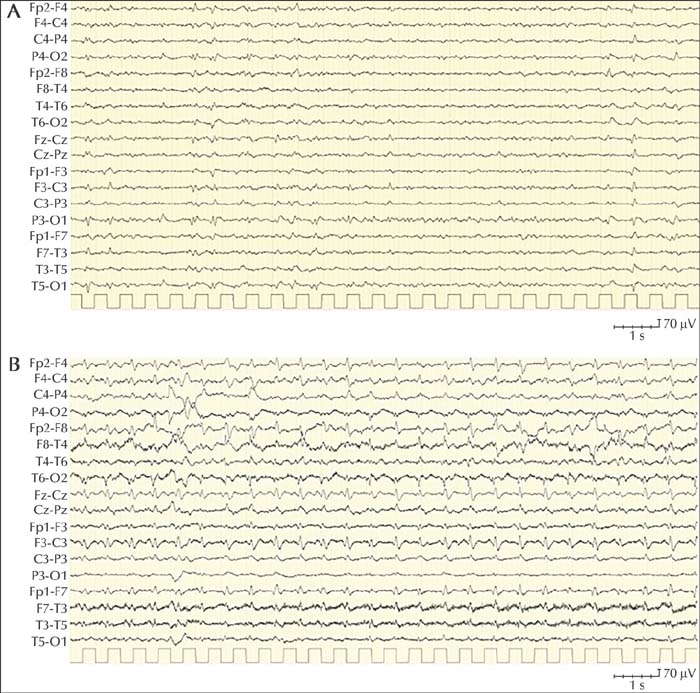
Published data on treatment show that most of the cases failed to respond to ASM and only a minority of subjects had a partial improvement (table 2). In particular, in most of the patients, non-convulsive status epilepticus was refractory to second and third lines of treatment (table 2). At present, no evidence is available to indicate which ASM is the most effective, however, due to the high prevalence of focal seizures, levetiracetam and lamotrigine are the drugs most often used. A 69-year-old woman without any significant antecedent and without a family history of neurological disorders was admitted to the hospital because of progressively worsening difficulties of speech, loss of fine motor dexterity in the right arm, unstable gait and difficulties in maintaining an upright posture without support. Her symptoms had started two weeks prior to admission. A CT scan was unremarkable. The EEG at admission showed slowing of the background activity with some diffuse sharp slow waves (figure 2A). Five days after hospitalization, she started to present with multiple daily episodes characterized by left head turning, stiffening of all four limbs and left limb clonic jerking. These episodes, initially stopped by administration of midazolam IV, recurred in the following days, sometimes spreading to the right limbs. An EEG 10 days after the first EEG, showed diffuse 1-2-Hz periodic sharp-wave complexes (figure 2B). Administration of midazolam IV did not modify the EEG. Chronic antiepileptic therapy with levetiracetam was undertaken with minimal and fluctuating improvement of seizures. Analysis of the CSF identified the presence of 14-3-3 protein (an non-specific marker of neuronal damage in prion disease). Real-time quaking-induced conversion (RT-QuIC) detected prion proteins in the CSF. These results prompted the diagnosis of probable sporadic CJD. During the following weeks, the patient developed global aphasia, afinalistic response to painful stimuli, head deviation to the left, right facio-brachio-crural hemiparesis, and dystonic head and left limb posture. She died three months after the onset of the symptoms. Case notes: The case described here is an example of probable sporadic Creutzfeldt-Jakob disease (according to the clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease) presenting with seizures a few days after the onset of the clinical symptoms. The patient presented with focal motor seizures, which are reported to be among the most common seizure types in prion diseases (see table 2). Seizures responded only partially to levetiracetam therapy.Case 2
Dementia with Lewy bodies
Dementia with Lewy bodies (DLB) is the third most common type of dementia after Alzheimer and vascular dementia [76]. Some studies show that the occurrence of seizures in DLB is higher than in the general population with an incidence rate of 2.62% for clinically diagnosed DLB and 3.8% for pathologically confirmed DLB [77]. A large retrospective analysis of seizures in patients with different types of dementia found a cumulative probability of developing seizures in DLB patients of 14.7%, which is higher than that observed in AD [17]. Age at seizure onset varies from 70 to 75 years and seizures may occur within four years before or after dementia onset [17]. The semiology of seizures is poorly reported, although a prevalence of focal or secondary generalized seizures has been observed [17]. As with Alzheimer, however, focal non-motor seizures may be undetected, hidden in the context of the neurocognitive disorder [17].
Regarding treatment, a study aiming to identify effective and safe ASMs in DLB patients concluded that the most suitable first-line drugs were lamotrigine, levetiracetam, lacosamide, brivaracetam, and gabapentin or pregabalin as second line [78].
Pathogenetic studies in animal models of DLB have demonstrated a role of alpha-synuclein in neuronal hyperexcitability [79]. In addition, pathological abnormalities frequently overlap between DLB and AD, such as neurofibrillary tangles and plaques, suggesting common epileptogenic mechanisms in these two conditions [80]. Indeed, the DLB-AD patient group displayed the highest prevalence of seizures (20.7%) [11], and synergistic interactions between the two disease processes have been previously demonstrated in double transgenic mice expressing human synuclein and APP [81].
Fronto-temporal dementia
A retrospective analysis revealed a cumulative probability of developing seizures in patients with frontotemporal dementia (FTD) of 3.0%, including also patients with progressive supranuclear palsy and cortico-basal degeneration as well as behavioural and linguistic variants of FTD [14]. FTD with parkinsonism linked to chromosome 17 (FTD-17q) has also been associated with epilepsy [82].
Mutations within the tau gene have been shown to cause FTD, demonstrating that tau dysfunction, in the absence of amyloid pathology, can be sufficient to cause neuronal loss and clinical dementia. In tau-negative FTD, TAR DNA-binding protein (TDP43) was identified as the major hallmark of this condition and the aggregation and cytoplasmic translocation of this nuclear protein are considered to contribute to the pathogenesis of FTD syndromes [83].
Tau protein, a hallmark of FTD pathology, has been suggested to play a role in epileptogenesis by modulating neuronal excitability in animal models of AD [84], and the presence of CSF total tau (T-tau) has been associated with a higher risk of developing seizures in humans [85].
Cortico-basal degeneration
A single case report of focal motor seizures in a 65-year-old patient with cortico-basal degeneration (CBD) exists in the literature. No alternative causes were found for her seizures, and the patient responded optimally to levetiracetam [86].
Parkinson disease
The association between Parkinson disease (PD) and epilepsy has always been considered very rare; almost “mutually exclusive” [87]. However, a recent retrospective study investigating the incidence of epileptic seizures in PD concluded that incident PD is associated with an increased risk of incident epileptic seizures [88]. After adjusting for potential confounding factors, a 1.7-fold increased risk of epileptic seizures in patients with PD compared with PD-free individuals was still observed. In particular, the risk was higher in PD patients with comorbid brain disorders, dementia, or more than one seizure provoking comorbidity. These findings are consistent with a cross-sectional study, which reported a prevalence ratio of PD of 3.19 (95% CI: 52.44-4.18) in patients with epilepsy aged more than 16 years, compared with non-epileptic individuals [89]. Interestingly, PD patients with comorbidities, that are known to be risk factors for epileptic seizures, had an even higher risk of epileptic seizures than patients without PD with such comorbidities [88]. Another study [90] reported a marked increase in the rate of SE in patients with idiopathic PD as compared to epilepsy patients, hypothesizing that the functional impairment of the basal ganglia in PD patients makes SE more likely by modulating thalamic nuclei involved in seizure maintenance, and thus suggesting a benefit from dopaminergic treatment in PD patients with SE [90]. An overview of published cases with Parkinson disease and epilepsy is presented in table 3.
A role of the basal ganglia in the propagation and control of epileptic seizures has been postulated [91] and, indeed, high-frequency transient stimulation of the subthalamic nucleus can suppress absence seizures in rats [92]. Further data from animal models and case reports suggest an antiepileptic effect by activation of dopamine receptor type 2, which is mainly under-stimulated in PD, and a potential protective effect of antiparkinsonian drugs on epileptic seizures [93]. Furthermore, zonisamide, an ASM with a dopaminergic effect, was reported to have beneficial effects on motor dysfunction and fluctuations in PD [94]. On the contrary, antipsychotic drugs that diminish dopaminergic transmission have been shown to facilitate seizures [93]. These data support the concept of a potential association between dopamine and epileptic seizures and emphasize the need for further research to investigate this relationship.
Progressive myoclonus epilepsies
Progressive myoclonus epilepsies (PME) represent a large group of neurodegenerative diseases associated with myoclonus, epilepsy and progressive neurological deterioration; in addition, photosensitivity, even at slow or single flash rates, can be a prominent clinical feature in some PME [94]. The majority are inherited in an autosomal recessive fashion, but autosomal dominant and mitochondrial inheritance are also known. PMEs account for less than 1% of all epilepsies. Age at onset varies from childhood to adult age depending on the specific disease, and the course is invariably fatal in most of these conditions [95].
PMEs comprise a large range of conditions including Unverricht-Lundborg disease, Lafora disease, neuronal ceroid lipofuscinosis, mitochondrial disorders (MERRF, POLG1, MELAS) and sialidosis. An overview of the main clinical and laboratory features of the various PMEs is reported in table 4 and in [95, 96]. In the following section, we summarize the main phenotypic features, pathogenetic mechanisms and treatment approaches for the most common PMEs.
Unverricht-Lundborg
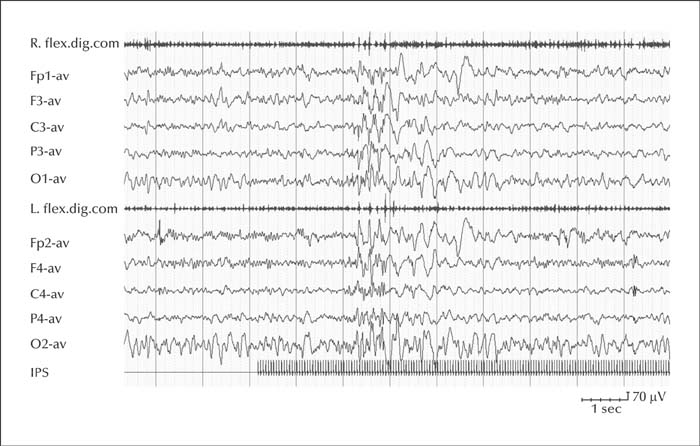
Unverricht-Lundborg disease (ULD) is the most common cause of PME and is due to a massive downregulation of the CSTB gene (or EPM1) that encodes for cystatin B [95]. It is considered the least severe form of PME, and in the initial stages of the disease, it can be misdiagnosed as juvenile myoclonic epilepsy [95]. Onset is between 7 and 13 years of age with progressive, stimulus-sensitive action myoclonus, tonic-clonic seizures and absence seizures. By six years after onset, myoclonus tends to worsen and patients develop ataxia and mild cognitive decline. Epilepsy tends to stabilize or even improve in early adulthood, although severely disabling stimulus-sensitive myoclonus and action myoclonus persist. EEG presents with background slowing and marked ictal and interictal generalized spike-waves discharges [95] (figure 3). Photic stimulation may facilitate spike-wave discharges but photosensitivity tends to abate and disappear after 10-15 years [95]. Valproate and levetiracetam are currently considered the most effective medications [95]. In most patients, seizure persistence and uncontrolled myoclonus require polytherapy with multiple ASMs [95]. The pathogenesis of seizures is not fully delineated. Experimental evidence from Cstb knock-out mice has shown that altered expression of cystatin B increases the activity of the protease, cathepsin B, which has the potential to damage neuronal function leading to hyperexcitability of cortical neuronal networks [95]. In addition, Cstb-deficient mice display increased susceptibility to kainate-induced seizures and display more neurodegeneration than controls, with loss of GABAergic hippocampal neurons resulting in defective GABAergic inhibition that leads to hyperexcitability of cortical neuronal networks and seizures [95].
Lafora disease
Lafora disease (LD) is due to autosomal recessive loss-of-function mutations in either the EPM2A gene (which encodes for laforin) or EPM2B gene (which encodes for malin) [95]. LD is a glycogen storage disease of which the pathological hallmark is “Lafora bodies” that consist of deposits of polyglucosan, an abnormal type of glycogen [96]. Onset of LD is between 8 and 18 years of age in previously normal adolescents, and is accompanied by cognitive decline, cerebellar signs, visual impairment, stimulus-sensitive myoclonic jerks and generalized seizures. Focal seizures with prevalent visual symptoms are also present. Seizures are drug-resistant and the condition progresses within 10 years to profound dementia and eventually to a vegetative state and death [95].
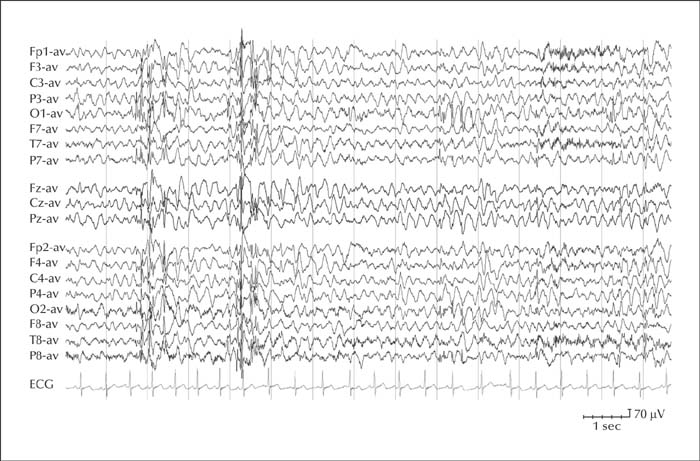
The EEG may be normal in the very early stages of the disease, then background activity rapidly slows down, generalized interictal spike-wave and polyspike discharges are elicited by photic stimulation, and posteriorly predominant multifocal epileptiform abnormalities appear (figure 4). Generalized discharges become almost continuous in the final stages of the disease [95].
A few ASMs have been shown to act on seizures and myoclonus, but none of them have been shown to affect the course of the disease. Valproate can, at least initially, suppress generalized seizures and photosensitivity, and topiramate and zonisamide may have antimyoclonic effects [95]. A good response to perampanel has also been reported [95].
As for pathogenesis, accumulation of Lafora bodies (LB) in neuronal dendrites could explain the cortical hyperexcitability in LD [95]. Several studies in cellular and animal models have shown that the malin-laforin complex is essential for many processes including glycogen metabolism [95]. Disruption of normal glycogen metabolism can cause hyperexcitability and epileptic seizures by compromising the glycogenolysis-dependent reuptake of extracellular K+ by astrocytes, thereby leading to increased extracellular K+ and associated membrane depolarization [95].
Neuronal ceroid lipofuscinosis
Neuronal ceroid lipofuscinosis (NCL), also known as Batten disease, comprises a group of neurodegenerative lysosomal storage disorders resulting in excessive accumulation of lipopigments [96]. NCLs are characterized by progressive decline of cognitive and motor functions, retinopathy with evolution to blindness, varying degrees of brain atrophy, and myoclonic epilepsy [95]. There are 14 forms of neuronal ceroid lipofuscinosis, classified according to age of symptom onset (varying from infancy and childhood to adulthood) and causative gene mutation [96].
ASMs commonly used are lamotrigine and valproic acid. Topiramate and levetiracetam can be also effective while carbamazepine, phenytoin and gabapentin may worsen myoclonic seizures [95].
Infantile NCL is a severe disease of infancy, which presents with seizures, developmental arrest and regression, and visual loss. The main gene involved in this form is CLN1, which encodes a lysosomal palmitoyl protein thioesterase. More severe than CLN1 is CLN10, which is a congenital fatal condition characterized by encephalopathy with respiratory insufficiency and status epilepticus. CLN10 encodes the lysosomal protease, cathepsin D [96].
Late-infantile NCL presents with onset in early childhood and is caused by pathogenic variants of the CLN2 gene. This disorder is characterized at the onset by a very severe myoclonic epilepsy, followed during the course of the disease, by cognitive and motor decline, and later by visual loss. CLN2 encodes a lysosomal tripeptidyl peptidase. Other NCLs such as CLN5, CLN6, CLN7, CLN8, and CLN14 mimic, to various extents, the clinical phenotype of the classic CLN2. Mild CLN6 mutations are another cause of adolescence or adult-onset PME [96]. CLN6 encodes an endoplasmic reticulum protein of unclear function.
The main features of juvenile NCL (CLN3) are onset in later childhood with retinal pathology and visual loss which are often the only symptoms for some years. Neurocognitive and motor decline eventually appear as well as seizures and myoclonus, the latter two present in a mild form for long periods. CLN3 encodes a lysosomal membrane protein with multiple attributed functions, including palmitoyl protein desaturase activity.
The onset of CLN4 (with mutation in the DNAJC5 gene; the only autosomal dominant NCL), CLN11 [96] and CLN13 (with mutation in the CTSF gene encoding lysosomal cathepsin F) occur in adulthood. CLN11 (with mutation in the GRN gene encoding the progranulin protein), similar to CLN3, presents with a prolonged period of severe visual impairment before the appearance of the PME phenotype [96]. Interestingly, other GRN mutations underlie completely distinct non-PME neurodegenerative disorders, such as frontotemporal dementia with TDP43-positive inclusions. CLN12 (with mutation in the ATP13A2 gene) can present with variable phenotypes, including teenage-onset PME, with some levodopa-responsive extrapyramidal features and one type of juvenile-onset Parkinson disease (PARK9; Kufor–Rakeb disease) [96]. CLN13 does not present as a PME since it is essentially a neurodegenerative disease with ataxia and dementia [96].
Concluding remarks
Epilepsy can be a comorbidity of some of the most common neurodegenerative pathologies and as the global population ages, the intersection between ageing, epilepsy and neurodegenerative disorders will become an increasingly pressing concern. Nevertheless, it is surprising to see to what extent epilepsy in neurodegenerative disorders remains underestimated. Studies in the elderly have supported the concept of a bidirectional association between dementia and epilepsy: indeed, not only do people with a dementing illness such as AD have an increased risk of subsequent epilepsy, but people with epilepsy are at a substantially increased risk of developing dementia.
Current evidence suggests that a process beginning long before the onset of clinically apparent seizures can potentially drive both epileptogenesis and cognitive decline. Yet, the role of epileptogenesis, either as the underlying culprit or as a consequence of neurodegeneration, remains unclear. Future avenues of research should clarify these mechanisms and investigate whether novel pharmacological therapies that target neurobiological pathways underpinning neurodegenerative diseases might potentially have anti-epileptogenic and disease-modifying effects on the seizures as well as on the progressive neurocognitive deterioration.
Neurologists will probably deal with these diseases to a growing extent in the future and understanding the epidemiology and the pathophysiology of epilepsy and degenerative disorders, particularly those affecting the elderly, might help to shape health care policies and reduce the burden of disease.
Supplementary material
Summary slides accompanying the manuscript are available at www.epilepticdisorders.com.
Disclosures
None of the authors have any conflicts of interest related to this study to disclose.Key points