Epileptic Disorders
MENUEpilepsia partialis continua associated with the p.Arg403Cys variant of the DNM1L gene: an unusual clinical progression with two episodes of super-refractory status epilepticus with a 13-year remission interval Volume 24, issue 1, February 2022


Dynamin-1-like (DNM1L) is a gene located on chromosome 12p11.21 that encodes for dynamin-related protein (DRP1), a GTPase involved in mitochondrial and peroxisomal fission and fusion, which plays a pivotal role in brain development [1]. Mice carriers of homozygous DNM1L deletion die during embryogenesis, while the heterozygous missense variant p.Arg403Cys causes reduced oligomerization, mitochondrial fission activity and mitochondrial recruitment of DRP1 [2].
Only 13 patients with this variant are described in the literature [3-11], presenting with a similar clinical course: a stormy onset and super-refractory status epilepticus during childhood (mean onset age at 6.6 years; range: 3-13 years), triggered in 60% of cases by infectious events or vaccination. The neurological outcome is generally extremely poor and brain MRI reveals recurrent features such as thalamic T2-FLAIR hyperintensity (50%) and progressive cerebral atrophy (80%).
We report the case of a 20-year-old female patient with this p.Arg403Cys mutation who showed biphasic disease progression, thus expanding the phenotypic spectrum associated with this mutation. The patient presented with two episodes of super-refractory focal myoclonic status epilepticus (the first at the age of six and the second at the age of 19), both with features of epilepsia partialis continua (EPC) with a 13-year interval of well-documented neurological and neuropsychological recovery, despite partial myoclonic seizures control.
Case study
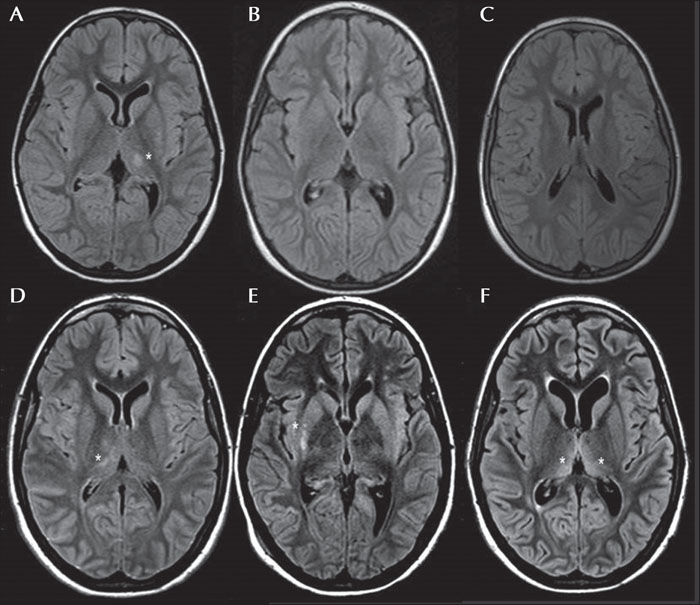
The patient was a right-handed Caucasian female with an uneventful birth and appropriate development. At the age of six, four days after having scarlet fever and 30 days after the second dose of the measles-mumps and rubella vaccine, she suddenly developed a focal motor status epilepticus characterized by non-resolving twitching of the right half of the face and right arm, with subsequent awareness impairment and coma. She was initially treated with high doses of intravenous benzodiazepines without benefit. She was then intubated and thiopentone and phenobarbital infusion was started, and a burst suppression was obtained on EEG. At the same time, the patient presented with hyperpyrexia and a measle-like rash. Inflammatory index, infectious investigation and lactate in the cerebrospinal fluid were negative. She was treated with ceftriaxone and acyclovir, with IV Immunoglobulins and steroids without benefit. In the acute setting, MRI showed transient T2 FLAIR hyperintensity on the left posterior thalamus and putamen without contrast enhancement (figure 1A). The super-refractory status lasted for 10 days with persistent myoclonic phenomena (right face and upper limb), until phenytoin was added to phenobarbital. She was then extubated, reaching a minimally conscious state. She showed restlessness, right hemiplegia and persistent right-arm myoclonic phenomena. Daily focal myoclonic seizures (right half of the face and arm) with awareness impairment, cyanosis and excessive drooling were also present.
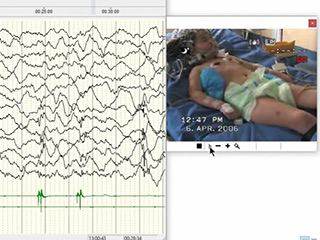
Continuous video-EEG with polygraphy revealed focal quasi-periodic (1-Hz) polyspike-and-wave discharges located on the left fronto-temporal regions (figure 2A, B). Evoked potentials from visual stimulation revealed increased latency that was more evident for the right stimulus, while somatosensory stimulation for both the upper and lower limbs did not show giant evoked potentials; nerve conduction velocity, electromyogram and C-reflex were normal. The electroclinical findings fitted with EPC (video part 1).
During hospitalization and in the following months, she was referred for several diagnostic investigations: genetic-metabolic (karyotype, subtelomeric rearrangement analyses, genetic analysis related to porphyria, MERRF, MELAS and POLG, urinary organic acids, long-chain fatty acids, and blood lactate concentration during clinical exercise), autoimmune (lupus anticoagulant antibodies, rheumatoid factor and anti-extractable nuclear antigen, anti-double-stranded DNA, anti-cardiolipin, anti-phospholipid, anti-mitochondria, anti-nuclear, and smooth muscle antibodies), infectious (MeV, RuV, EBV, CMV, HSV1, HSV2, HSV6, enterovirus, parvovirus, and Borrelia), and ophthalmologic evaluation; all without significant results.
Twenty months later, the control MRI confirmed the complete resolution of the FLAIR hyperintensity (figure 1B, C). A brain PET evaluation and MRI spectroscopy were negative.
A muscle biopsy (for histology and enzymology) was performed based on a suspicion of mitochondrial aetiopathology, without any pathognomonic features.
In the following 13 years, she showed a progressive improvement in her clinical condition. Despite residual moderate intellectual disability (full intellectual quotient: 44), she developed left-handed manual preference and lecture skills, and she achieved a high-school diploma with teaching assistance. She displayed a significant reduction of myoclonic phenomena, which were only triggered by spontaneous movements and/or emotional states. She had rare (three to four episodes/year) and isolated focal right-side clonic seizures, which were notably catamenial. Jerk-locked back averaging confirmed the left central cortex as the origin of the epileptic myoclonus, although we could not exclude the coexistence of widespread subcortical myoclonus [12]. Several AEDs, such as clonazepam, clobazam, carbamazepine, levetiracetam, perampanel and topiramate were administered over the years with a significant progressive disappearance of the EEG abnormalities (figure 2C, video part 2).
At the age of 19, without any noticeable trigger, she suddenly presented with convulsive super-refractory status epilepticus, with myoclonic jerks of the left arm (figure 2D). She was treated with thiopentone, propofol, phenobarbital, phenytoin and the ketogenic diet, with only partial resolution of the status. At onset, brain MRI showed a transient multifocal T2-FLAIR hyperintensity signal in the right capsular, thalamic, posterior lentiform nucleus, posterior frontal areas, as well as in the centro-parietal and mesial frontal cortex (figure 1D, E and video part 3). MRI after two months showed progressive bilateral atrophy with ventricular enlargement and resolution of the right cortical and posterior lenticular hyperintensity. Residual bilateral dorso-medial thalamic hyperintensity, probably related to the prolonged status, was observed (figure 1F). A consequential vegetative state, with persistent epileptic and non-epileptic myoclonus with face and upper limb involvement (mainly visible on the left side), endured.
Eight months later, concurrent with pneumonia, convulsive status worsening was observed which was treated with propofol, ketamine, midazolam, thipentone, lamotrigine, zonisamide, levetiracetam, perampanel, lacosamide, clonazepam, IV Immunoglobulins and steroids (figure 2E). A second trial of the ketogenic diet was performed without obtaining adequate ketoacidosis. Sodium valproate was therefore introduced with partial improvement regarding the myoclonic features, without hepatic side effects. Meanwhile, the proband and her parents were referred for whole-exome sequencing (Sure Select Human All Exon V7, Agilent) which revealed a de novo missense variant (c.107C>T, p.Arg403Cys) in the DNM1L gene which is considered to be pathogenic based on the literature.
The patient is now 20 years old and she currently manifests with a persistent vegetative state with drug-resistant epileptic and non-epileptic myoclonus, revealed by EEG polygraphy, only during awake recordings, characterized by prevalent right hemiface and upper limb involvement (figure 2F, video part 4) [13]. Piracetam was recently introduced in addition to clonazepam, lacosamide, lamotrigine, levetiracetam, perampanel and sodium valproate, with only partial benefit.
Discussion
Only 13 patients with the p.Arg403Cys DNM1L missense mutation have been described and the disease spectrum is only beginning to be clarified. Adding our patient to all the reported cases, we confirm that the most frequent clinical phenotype seems to be characterized by childhood-onset (mean age: 6.5 years) status epilepticus in 65% of cases, related in time to infectious episodes or vaccination. Based on the extensive (15-year) follow-up with yearly visits, our case has peculiar characteristics regarding both prominent EPC features [14] and prolonged improvement based on neurological and video-EEG examination, which has not previously been described. Therefore, this missense variant could be considered as a cause of EPC.
In addition, the prevalent MRI features described were transient subcortical thalamic T2-FLAIR hyperintensity (54%) at onset, followed by progressive cerebral atrophy (81%). During both episodes of status, migrating transient thalamic FLAIR hyperintensity (left-sided during the first status and right-sided during the second) was detected. These neurological data were also congruent with the side of clinically observed myoclonic phenomena, which reverted during the remission interval. It would be interesting to evaluate whether these neuroradiological features associated with EPC could be considered a precocious marker of the disease, or whether they are merely the consequence of status epilepticus, considering that these MRI features are most prevalent in mitochondrial disorders.
Epigenetic and/or therapeutic protective factors that could account for this different evolution also remain unidentified. It has been recently outlined that this mutation can clinically simulate an immune-mediated encephalopathy [9]. Due to the role of mitochondria in the respiratory chain and inflammatory response in regulating cell death and mitophagy, this missense variant is likely to be involved in oxidative damage and the inflammatory process. However, all the investigations were not clearly pathognomonic for mitochondrial encephalomyopathy.
In our patient, the presence of a minor illness and/or vaccine at onset and the prolonged period of remission lead us to hypothesize that an unknown immune-modulating factor could have affected the course and prognosis of the disease, more than the AEDs. Several therapeutic options were tried over the years, however, no drug in particular significantly changed the disease course. During the second episode of status, only high-dose clonazepam (10 mg/die, 0.2 mg/kg/die) provided benefit for myoclonic phenomena [15].
In conclusion, we would like to highlight the main features of our patient as EPC and super-refractory status epilepticus, with transient and migrating subcortical thalamic MRI hyperintensity at onset. Moreover, DNM1L encephalopathy may apparently display a prolonged remission period with satisfying seizure control, however, persistent cognitive impairment may ensue, which could distract clinicians from the progressive evolution and the genetic bases of this encephalopathy. Precise seizure characterization and whole-exome sequencing in patients with childhood-onset super-refractory status are crucial in order to establish early diagnosis.
Supplementary material
Summary slides accompanying the manuscript are available at www.epilepticdisorders.com.
Disclosures
None of the authors have any conflicts of interest to declare.
Funding sources
The work was supported by funding from the Ministero della Salute (Italia), Grant # RC2020 2021 and 5xmille funds (to CZ, MTB).