Epileptic Disorders
MENUCrossing the lines between epilepsy syndromes: a myoclonic epilepsy variant with prominent eyelid myoclonia and atonic components Volume 20, issue 1, February 2018




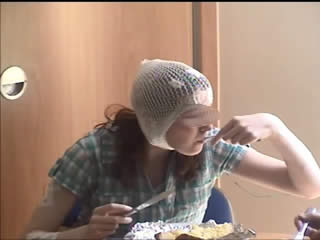
Effective classification of epileptic seizures and syndromes is essential for appropriate management and prognosis. Syndromic diagnosis is based on well-defined clinical and EEG features that differentiate distinct nosological entities with individual natural history. In clinical practice, however, there are patients with unusual manifestations whose electroclinical features cannot fit neatly into a recognized epilepsy syndrome or cases where syndromes appear to overlap, posing a diagnostic challenge. This heterogeneity has been documented in the literature in several cases marked by the presence of eyelid myoclonia with or without absences (EMA) in which either a diagnosis of a Jeavons syndrome (JS) variant was proposed (Sevgi Demirci and Saygi, 2006; Capovilla et al., 2009; Nar Senol et al., 2015) or EMA was considered an electroclinical feature of a distinct epilepsy syndrome (Caraballo et al., 2009).
Jeavons first described this entity in 1977 (Jeavons, 1977) and thereafter it has been confirmed by several authors (Appleton et al., 1993; Giannakodimos and Panayiotopoulos, 1996; Striano et al., 2002) as a well-defined epilepsy syndrome of childhood, classified as a genetic generalized epilepsy (GGE), although not yet included in the official ILAE position papers (Engel, 2001; Berg et al., 2010). It is characterized by:
- –eyelid myoclonia with or without absences;
- –eye closure-induced seizures, EEG paroxysms, or both;
- –and photosensitivity.
Generalised tonic-clonic seizures (GTCS) are not uncommon in its clinical course, but the presence of myoclonic seizures other than eyelid myoclonias (EM) is a matter of debate (Panayiotopoulos, 2005; Striano et al., 2009). There have been case descriptions in the literature, referred to as JS variants, with massive myoclonus, intellectual disability, or slowing of the EEG background and cases of dynamic overlap with other genetic generalized syndromes such as juvenile myoclonic epilepsy (JME) (Sevgi Demirci and Saygi, 2006; Yalçin et al., 2006; Capovilla et al., 2009; Nar Senol et al., 2015). Moreover, recently there has been evidence of the possible role of the occipital cortex in initiating the generalized epilepsy network in JS (Viravan et al., 2011).
Adding to the phenotypic heterogeneity of GGEs, we describe two cases of paediatric patients who exhibited the unusual clinical association of myoclonic epilepsy with prominent eyelid myoclonias, atonic components, and cognitive impairment. We also note the presence of interictal and ictal posterior discharges during eyelid myoclonia in one of these two patients, in support of similar previous observations.
Case 1
A 15-year-old female with drug-resistant epilepsy was referred for a three-day video telemetry study to capture and analyse seizures with the aim of diagnosing her epilepsy syndrome. At the age of 11 years, she presented with an episode of GTCS that occurred after a stressful week at school. On assessment, she reported having had myoclonic jerks for at least 18 months previously. The myoclonic jerks were occurring daily, especially on awakening, involving predominantly her left upper arm. She was diagnosed with juvenile myoclonic epilepsy (JME) and was started on lamotrigine, which was later substituted with valproate due to worsening of seizures and mood deterioration. The jerks gradually increased over time, especially immediately prior to the occurrence of the presenting tonic-clonic seizure. Her first EEG revealed bursts of generalized polyspike-waves with evidence of photosensitivity. Her MRI brain scan was normal. Her mother had a history of two tonic-clonic seizures at a younger age (but did not require treatment) and her younger brother also had an episode of GTCS and is undergoing investigation. She was born at 42 weeks, her motor development was within normal limits but her language and communication skills were delayed and she was diagnosed with autism spectrum disorder and learning difficulties at the age of 12 years. She had also previously experienced depressive symptoms.
During a follow-up EEG, eyelid myoclonia with absences was first recognised as a “new” seizure type (in hindsight, she had been having these eyelid movements and episodes of daydreaming from early childhood, but they had not been considered as epileptic events).
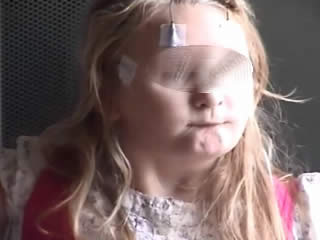
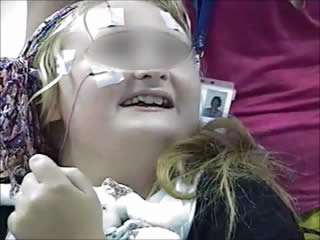
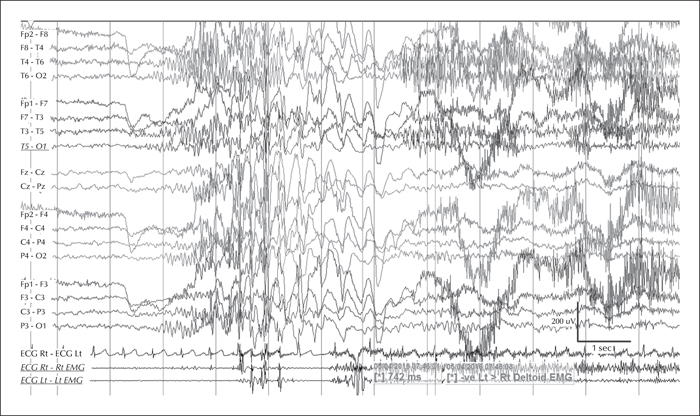
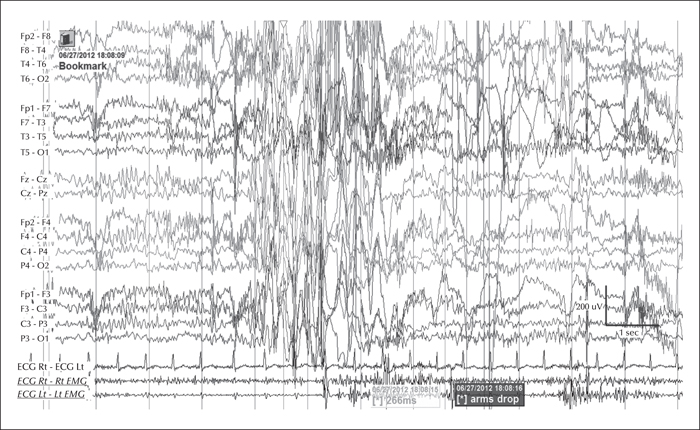
The video telemetry showed normal background with a well formed alpha rhythm at 9-12 Hz which could occasionally have a spiky contour, with prominent posterior discharges on eye closure, and frequent eyelid myoclonias (video sequence 1) with and without myoclonic jerks, predominantly affecting her left upper limb (video sequence 2). Retropulsion of the head was also noted (video sequence 3) with evidence of photosensitivity. All these events were associated with an initial run of posterior spiky discharges, which became more generalised with polyspikes and intermixed slow waves. Furthermore, definite atonic events, lasting up to 742 ms (video sequence 4), were noted on EMG (figures 1 and 2). She was on sodium valproate at the time of the recording.
Case 2
A 12-year-old female with a longstanding history of refractory epilepsy was referred for video telemetry to document seizures. Her first paroxysmal events started at the age of 3 years, consisting of brief eye rolling episodes of her right eye which had been witnessed once every month. Episodes increased in frequency over the years, with both eyes rolling upwards and eyelid flickering, related to changes in brightness levels. During these episodes, her head would occasionally drop to one side and tilt backwards, and at times, she would become withdrawn and unsteady for a few seconds with glazed eyes and slurred speech. By the age of 6 to 7 years, she had also developed frequent head nods and myoclonic seizures involving her head, shoulders, and trunk, occurring singly or in clusters. An EEG at the time showed bursts of atypical generalized spike and wave activity at 2 Hz associated with head twitching and evidence of photosensitivity. Her MRI brain scan was normal and her family history was unremarkable.
She was started on antiepileptic treatment but her seizures proved refractory to several appropriate AEDs and she also developed serious adverse effects, which led to her being taken off all medication. Ketogenic diet therapy was trialled for six months but was not successful in controlling her seizures. Vagal nerve stimulation was also unsuccessful and was turned off at the time of assessment.
She was born at 42 weeks with induced labour. Her initial development was normal, but her educational progress and social communication skills plateaued at around the age of 5 years, and she was diagnosed with mild learning difficulties and anxiety disorder with challenging behaviour and aggression. On examination, she was felt to have some subtle dysmorphic features, but her genetic and neurometabolic investigations were normal.
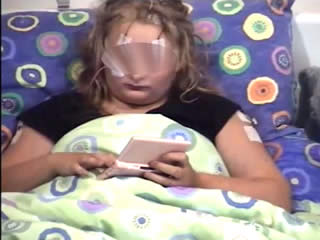
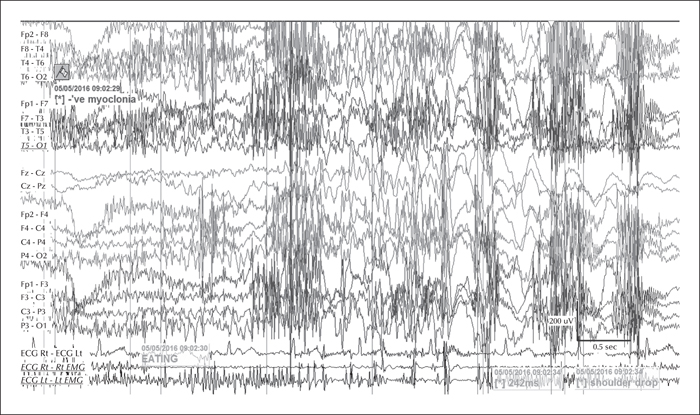
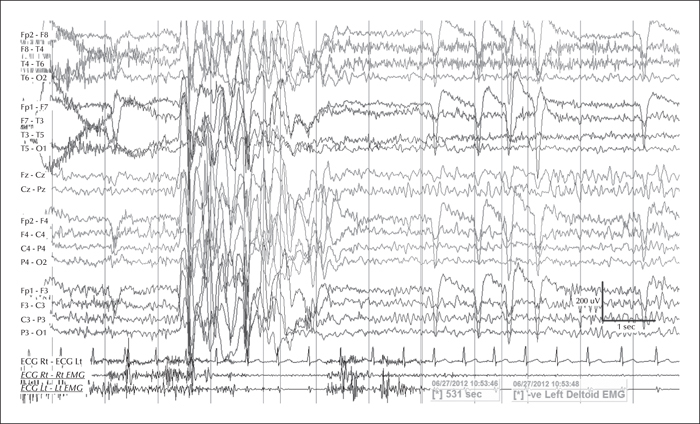
During the three-day video telemetry recording, she had frequent brief seizures, mostly eyelid myoclonia following eye closure (video sequence 5) and myoclonic jerks involving head, neck, shoulders, and arms (video sequence 6, 7). Head retropulsion (video sequence 7) was also noted. These were associated with bursts of generalised, but frontally dominant, irregular 2.5-3-Hz spike-slow-wave discharges. There was also evidence of atonia on EMG, lasting up to 531 ms (figures 3 and 4), associated with brief arm or head drops (video sequence 8). Awake background and sleep architecture were normal and no convincing photosensitivity was demonstrable at this time. She was not on any medications at that time due to the previous side effects.
Discussion
In both of these cases, the occurrence of eyelid myoclonias with and without absences, eye closure paroxysms, and evidence of photosensitivity in two female patients with normal neurological examination and brain imaging could be suggestive of Jeavons syndrome. Photosensitivity can be suppressed by antiepileptic medication and may diminish with age, as demonstrated in the case of the second patient. However, the striking myoclonic components seen in both girls are not typical of this epileptic condition and could be consistent with the diagnosis of JME, initially proposed in the first case. Furthermore, the presence of atonia in the form of brief head and arm drops recorded on video-EEG polygraphy is rather unusual for both of these conditions. All these features suggest either an atypical JS variant or an “overlap” GGE phenotype. Of note, the patients were not on any medications which could have induced these symptoms.
A few reports in the literature have described similar cases with unusual presentation of eyelid myoclonias with or without absences during the clinical course of childhood epilepsy, suggesting that their occurrence alone is not sufficient to characterize a definite epilepsy syndrome. Yalçin et al. (2006) reported four patients whose first seizure type at the age of 11 to 13 years was EMA and who later presented with myoclonic seizures of the extremities and were diagnosed with JME. Caraballo and colleagues studied 63 cases from Italy and Argentina and in almost one third of these, EMA was thought to represent a type of seizure occurring in patients with GGEs who also experienced massive myoclonus and/or GTCS (Caraballo et al., 2009). There were also cases where EMA occurred during the course of a cryptogenic myoclonic epilepsy, years after the occurrence of myoclonic seizures or even following the diagnosis of benign myoclonic epilepsy of infancy (Moutaouakil et al., 2010; Ohya et al., 2012). In a recent study of 61 adult cases (Nar Senol et al., 2015), patients with EMA were divided into three distinct groups; those who had pure JS with EMA as the predominant seizure type, a second group of JS variants with EM seizures associated with GTCSs and/or massive myoclonias, and a third group of patients with EMA who had epilepsy, previously referred to as a symptomatic epilepsy. Capovilla et al. described another variant among patients with EM, characterized by eyelid fluttering, typical EEG pattern, and impaired intellectual function (Capovilla et al., 2009), and a further case of EMA with mild intellectual disability and background slowing on EEG was highlighted by Sevgi Demirci and Saygi (2006). Both patients presented here had cognitive impairment, as well as features of autistic spectrum disorder, but to our knowledge, this is the first report of atonia being clinically associated with EMA in the setting of a myoclonic epilepsy.
In recent studies and based on case descriptions, it has been suggested that the occipital cortex plays a central role in the generation of eye closure-induced EMA, initiating a generalized epilepsy network which involves thalamocortical and transcortical pathways and the brainstem (Viravan et al., 2011). In support of this theory, we also documented prominent focal discharges arising from the occipital regions on the EEG of the first patient, both interictally and ictally during eyelid myoclonias, as an initial run of posterior spiky discharges which became more generalized. We also noted the presence of occasionally spiky posterior alpha activity in the same patient, as reported by Viravan et al. (2011) in their study.
Conclusion
Our description of atonic components enlarges the spectrum of seizure types that can be associated with eyelid myoclonia with and without absences, adding to the variability of Jeavons syndrome and GGEs “overlap” phenotypes. The occurrence of focal posterior discharges in one of our patients provides further evidence in support of the hypothesis considering EMA to be an occipital cortex-initiated generalized seizure activity.
Supplementary data
Summary didactic slides are available on the www.epilepticdisorders.com website.
Acknowledgements and disclosures
The authors are grateful to the patients and their families for their collaboration and support. UCL Great Ormond Street Institute of Child Health received funding as an NIHR Biomedical Research Centre.
None of the authors have any conflict of interest to declare.