Epileptic Disorders
MENUClinical evolution and epilepsy outcome in three patients with CDKL5-related developmental encephalopathy Volume 21, issue 3, June 2019

The association between cyclin-dependent kinase like 5 (CDKL5) mutations and a severe developmental and epileptic encephalopathy (EE) was first described in 2003 in a female with Rett syndrome (RS) (Kalscheuer et al., 2003). CDKL5 gene mutations have been also associated with West syndrome (Guerrini and Parrini, 2012). To date, CDKL5 disorder is considered as an independent clinical entity, associated with early-onset EE (Fehr, 2013). The diagnosis is suspected based on normal prenatal history, early-onset epilepsy, stereotypical hand movements, severely impaired psychomotor development, and hypotonia, and then confirmed by detecting heterozygous CDKL5 gene mutations. The attainment of developmental milestones is severely impaired in CDKL5 disorder. Clinical variability is described, and although milestones are attained, they are significantly delayed (Bahi-Buisson et al., 2008; Fehr et al., 2015). The natural history of epilepsy in CDKL5 patients is characterized by three stages: (1) early epilepsy; (2) EE with infantile spasms; and finally, (3) late multifocal and myoclonic epilepsy (Bahi-Buisson et al., 2008). These stages are often interrupted by “honeymoon periods”, a time when seizures become transiently responsive to treatment or spontaneously regress (reported as lasting 2-30 months) after early seizure onset (Bahi-Buisson et al., 2008; Guerrini and Parrini, 2012). Regardless of the honeymoon period, progressive developmental deterioration (DD) is persistent. This dichotomy in the course of CDKL5 epilepsy and psychomotor development is also consistent with previous reports (Bahi-Buisson et al., 2008; Guerrini and Parrini,2012; Fehr et al., 2015).
We describe the electroclinical findings and psychomotor development of three girls (table 1) with de novo CDKL5 mutations who showed apparent dissociation between epilepsy severity and evolution of DD, thus raising the question whether the developmental delay is the result of epileptic activity, or a consequence of the underlying cerebral pathology.
Clinical descriptions
Patient 1
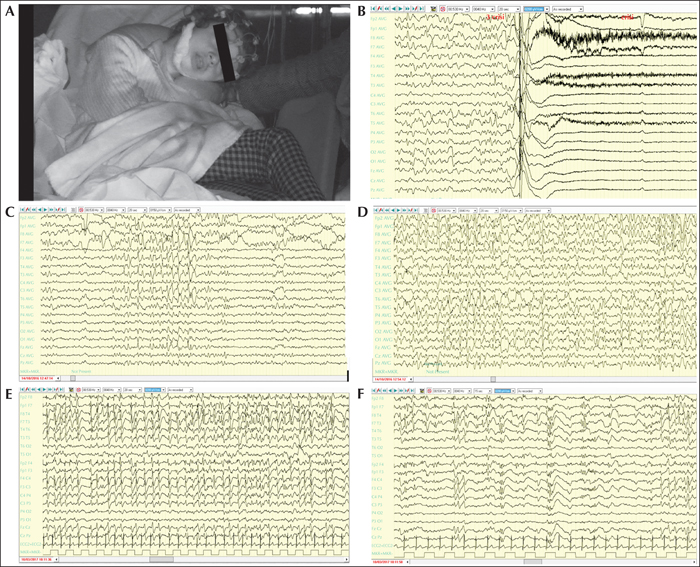
This 11-year-old girl has previously been reported (Artuso et al., 2010). The parents reported poor sucking and regurgitation in the neonatal period. At 30 days of age, she presented with repeated seizures, characterized by eye rolling, head deviation, generalized tremors, and autonomic signs, lasting about 30 seconds, several times a day. The EEGs showed spikes and slow waves over the temporal or centro-temporal areas, predominantly over the right side. Phenobarbital (PB) was started which led to a reduction in seizure frequency. At the age of 20 months, epileptic spasms appeared. Valproic acid (VPA) and vigabatrin (VGB) failed to control seizures, and she started therapy with ACTH. During the following years, she presented with various seizure types during sleep and wakefulness, including myoclonus of the upper limbs, episodes of cyanosis and tonic seizures, and episodes of crying associated with a scared gaze, followed by tonic extension of the superior limbs. The EEG showed slow background activity and multifocal spike and sharp wave complexes over the frontal areas, which were worsened by sleep. She did not experience “honeymoon periods” but only transitory periods (of less than one month) and slight improvement in seizure frequency due the introduction of new antiepileptic drugs (AEDs). Her psychomotor development was severely delayed; she never walked and never acquired language skills. At the age of two years, hand clapping and mouth-hand stereotypies appeared. At our first evaluation (aged 11 years), she was not able to hold her head and sit unsupported, visual contact was absent, and she had generalized hypotonia (figure 1A) and frequent seizures (more than 20/day). Some seizures were characterized by initial spasms, followed by rapid and multidirectional eye movements, lasting one to two minutes, accompanied by electro-decremental changes and generalized bursts of polyspike complexes on EEG (figure 1B). Other seizures were noted and characterized by sudden awakening and eye movements as well as tonic seizures. Interictal EEG showed slowing of the background activity with bilateral high-amplitude delta waves, intermixed with generalized spikes and polyspikes and an absence of physiological sleep figures (figure 1C, D).
Sanger sequencing showed the presence of a pathogenic CDKL5 mutation, c.1039C>T (p.Q347X) (NM_003159.2), rs267608561, affecting the C-terminal regulatory domain, due to maternal germinal mosaicism. The mutation has been reported to be responsible for early-infantile epileptic encephalopathy 2 in one patient based on the ClinVar database (SCV000188322.2) (Landrum et al., 2018). Sadly, we do not have more clinical information about the phenotype of this patient.
Patient 2
This was a 10-year-old girl; the first daughter of healthy unrelated parents. Psychomotor development was severely delayed; she never spoke nor reached an upright position. At the age of 2.5 years, she showed mouth-hand stereotypies, autistic features, and sleep disorders. At seven months of age, she presented with epileptic spasms. At this age, psychomotor development was already delayed (she had not acquired head control or the sitting position). Her EEG showed generalized bursts of polyspikes, and spike and waves predominating over the central areas. She started therapy with ACTH and, after six months, VPA was added because of the worsening of seizures. At two years old, after a brief period of seizure freedom, seizures reappeared and the ketogenic diet (KD) was initiated. However, KD was withdrawn after a few months because of eating difficulties. Topiramate (TPM) and clonazepam (CLN) were added. From the age of three to seven and a half, the patient experienced a long seizure-free interval. At this age, she started levetiracetam (LEV) because of tonic-clonic seizures. From the age of three years, she has been seizure-free. However, her EEG showed continuous spikes and slow waves and disorganized background with slowing and bifrontal spikes (figure 1E, F). At the time of our evaluation, at the age of 10, she had poor eye contact and an absence of response to social interactions and severe hypotonia. She was not able to hold her head and sit unsupported. Her EEG showed continuous spikes and slow-wave complexes without sleep organization, as in the previous patient. CGH-array analysis revealed a Xp22.13 de novo microdeletion (18,565616-18,588,650) (USCS Hg 19) spanning 23 Kb, involving exon 4 and intron 3 and 4 of the CDKL5 gene. This variant is not reported in Decipher as well as in the Database of Genomic Variants (DGVs).
Patient 3
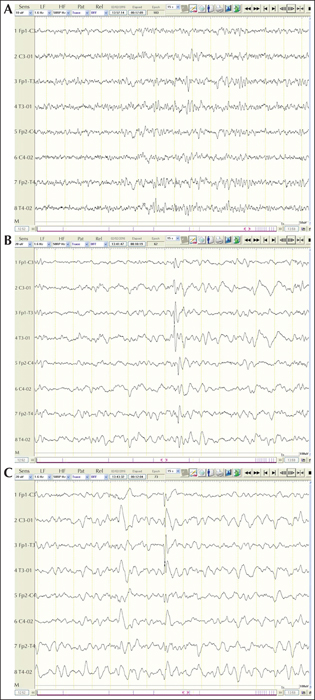
This six-year-old girl was the third daughter of healthy unrelated parents. From the first weeks of life, the parents reported irritability, feeding difficulties, and excessive crying. Her psychomotor development was severely delayed; she never walked nor acquired language skills. She showed hand apraxia, poor eye gaze, axial hypotonus, nystagmus, and acquired microcephaly. At the age of 18 months, she showed mouth-hand and hand clapping stereotypies. At 20 days of life, the girl presented with episodes of conjugate upward eye deviation and ocular clonus. The episodes occurred several times a day and PB was started with a reduction in seizures. EEGs showed moderately slow background activity with multifocal discharges. At the age of five months, she experienced seizures characterized by staring, followed by clusters of asymmetrical extension spasms lasting for a few minutes and upon awakening. At 12 months, because of a reduced seizure frequency and absence of EEG abnormalities, AEDs were gradually withdrawn, with no changes in the frequency and intensity of seizures. Furthermore, for six months, no seizures were observed. At two years of age, she developed focal seizures, usually during sleep, with a frequency of about three episodes per week. The interictal EEG showed mild background slowing and left frontal-temporal spikes or multifocal spikes (figure 2). VPA controlled seizures and she currently has one to three focal seizures per month. She is able to sit unsupported despite her generalized hypotonia, but she has never acquired independent walking or language. Targeted re-sequencing for epileptic encephalopathy genes revealed a de novo mutation in CDKL5 (c.173 del, p.Leu58TYrfsX18) not previously reported, which leads to a truncation of the protein within its catalytic domain.
Discussion
We describe the electroclinical and neurodevelopmental features of three girls with CDKL5 mutations. The key clinical feature of CDKL5 disorders is a severe and early-onset epilepsy (Bahi-Buisson et al., 2008; Fehr et al., 2015; Fehr et al., 2016). Epilepsy starts within five months with frequent seizures; spontaneous fluctuations in seizure frequency and a good response to AEDs are possible (30% of the series by Bahi-Buisson et al. [2008]). A longer duration of “honeymoon periods” clearly correlates with a better epilepsy outcome, while DD proceeds independently. In our cases, the dissociation between epilepsy course and the neurological impairment is consistent with previous reports (Bahi Buisson et al., 2008). All patients in the study of Bahi-Buisson et al. (2008) had severe DD at seizure onset, which persisted irrespective of their later epilepsy course. Also, in the case series of Muller et al. (2016), patients did not exhibit improvement in cognitive functions even if a significant reduction in seizures was achieved. Conversely, Fehr et al. (2015) suggested that functional abilities (the ability to walk and use spoken language) were associated with lower rates of current seizure frequency. The attempt to investigate a possible relationship between DD and genotype has not provided significant results, with the exception of a more favourable psychomotor development in females with a truncating mutation towards the end of the gene (Fehr et al., 2016). Other authors suggested that subjects with p.Ala40Val, a missense mutation within the ATP binding site of the kinase domain, had a milder phenotype than those with a missense mutation elsewhere within the kinase domain or a frameshift within the C-terminal region (Guerrini and Parrini, 2012; Fehr et al., 2016).
Our patients showed severe psychomotor and DD, apparently not related to the frequency of seizures or presence or absence of “honeymoon periods”. Patient 1 showed severe epilepsy from the first month of life, with a progressive increase in frequency of seizures that were drug resistant. Her EEG pattern was suggestive of a severe encephalopathy, consistent with the observed severe DD. Patient 2 and 3 displayed severe hypotonia, an absence of language and walking abilities, stereotypies, and autistic-like behaviours. However, their epileptic history was characterized by a long “honeymoon period” (three years for the second patient and six months for the third patient). The EEG pattern for the second patient was suggestive of a severe encephalopathy with a continuous epileptic activity without sleep figures. Control of seizures was discrete, but the global clinical status overlapped with that of other patients.
Some evidence from the literature suggests that a specific type of EE (as status epilepticus during sleep), observed, for example, in our second patient, is related to worse cognitive performance (Specchio et al., 2012; Howell et al., 2016). Other authors have also reported a severe EEG pattern even in the absence of seizures in patients with CDKL5 mutations. In this context, EEG for patients with CDLK5 disorder might be a useful marker for assessing neurological development (Bahi-Buisson et al., 2008), as in our second patient.
Early seizure onset is considered one of the most relevant risk factors for EE severity (Specchio et al., 2012; Nardou et al., 2013) and there is evidence that lower seizure frequency is associated with better functional and language skills in subjects with CDKL5 mutations (Fehr et al., 2016). Moreover, the use of antiepileptic polytherapies in the first years of life could worsen cognitive functioning in these children, especially if the drugs are not indicated for the specific epileptic syndrome, as emerged in the studies on Dravet syndrome (de Lange et al., 2018).
In our patients, epilepsy started later in Patient 2, who showed longer “honeymoon periods” compared with the other two individuals. Polytherapy was used in two of our patients, but this aspect should be investigated further with extensive case studies. No correlation emerged between seizure frequency and psychomotor development, thus suggesting that the type of genetic dysfunction is crucial to eventually determine the neurodevelopmental outcome. Moreover, it is difficult to determine the causative role of the underlying genetic dysfunction and that of the consequent seizures in influencing psychomotor outcome in children with EE (Striano et al., 2013). It remains to be determined whether the impairment seen in CDKL5 disorder is a direct result of the occurrence of infantile seizures, resulting in an EE, or whether it is a consequence of the underlying gene mutation. According to Muller et al. (2016), in CDKL5 disorder, the EE should be considered as part of the syndrome, thus the epilepsy should not be considered as the cause of severe neurological impairment. Concerning the role of EEG data, age at seizure onset, seizure frequency, and duration of the honeymoon period during the natural history of CDKL5 gene-related developmental encephalopathy, data in the literature are scarce. These aspects could be related to the small samples in previous studies that do not allow conclusions to be drawn. The correlation between epilepsy and psychomotor development is an intriguing and complex topic as an evolving brain susceptible to continuous changes is affected. It is important to further examine the variability of CDKL5-related disorder so that appropriate prognostic information can be provided. The description of our patients may be useful for further studies aiming to investigate the relationship between the epilepsy and neurodevelopmental aspects of CDKL5-related disorder, as in other genetic epilepsies.
Acknowledgements and disclosures
We thank the patients’ families for giving us permission to report their medical history.
None of the authors have any conflict of interest to declare.